

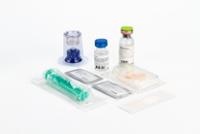
KYBERNIN P 500 UI Pó e Solvente para Solução Injectável ou Perfusão
Pergunte a um médico sobre a prescrição de KYBERNIN P 500 UI Pó e Solvente para Solução Injectável ou Perfusão



Como usar KYBERNIN P 500 UI Pó e Solvente para Solução Injectável ou Perfusão
Introdução
Prospecto: informação para o utilizador
Kybernin P 500 UI pó e dissolvente para solução injectável e para perfusão
Antitrombina III humana
Leia todo o prospecto atentamente antes de começar a usar este medicamento, porque contém informações importantes para si.
- Conserva este prospecto, porque pode ter que voltar a lê-lo.
- Se tiver alguma dúvida, consulte o seu médico ou farmacêutico.
- Este medicamento foi-lhe prescrito apenas a si, e não deve dá-lo a outras pessoas, mesmo que tenham os mesmos sintomas que si, porque pode prejudicá-las.
- Se experimentar efeitos adversos, consulte o seu médico ou farmacêutico, mesmo que se trate de efeitos adversos que não aparecem neste prospecto. Ver secção 4.
Conteúdo do prospecto
- O que é Kybernin P e para que é utilizado
- O que necessita de saber antes de começar a usar Kybernin P
- Como usar Kybernin P
- Posíveis efeitos adversos
- Conservação de Kybernin P
- Conteúdo do envase e informação adicional
1. O que é Kybernin P e para que é utilizado
Kybernin P é um pó e dissolvente para solução injectável e para perfusão.
Este medicamento pertence ao grupo de medicamentos chamados agentes antitrombóticos.
Kybernin P é utilizado se si tem um défice congénito de antitrombina, para prevenir a formação e o desenvolvimento de coágulos nos vasos sanguíneos das suas pernas (trombose venosa profunda) ou noutros vasos do seu corpo (tromboembolismo) durante a cirurgia ou no período peri-parto e em associação com heparina se estiver indicado.
Kybernin P também é utilizado se si tem défice adquirido de antitrombina.
2. O que necessita de saber antes de começar a usar Kybernin P
Não use Kybernin P:
Se é alérgico ao princípio ativo ou a algum dos outros componentes deste medicamento (incluídos na secção 6).
Advertências e precauções
Assim como com qualquer produto proteico para administração intravenosa, é possível que se produzam reações de hipersensibilidade de tipo alérgico. É necessária uma estreita monitorização e observação cuidadosa dos doentes para detectar qualquer sintoma durante o período de perfusão. É necessário informar os doentes sobre os sinais iniciais das reações de hipersensibilidade, que incluem erupções cutâneas que podem chegar a urticária generalizada, opressão torácica, dificuldade em respirar, hipotensão e anafilaxia. Se se produzirem estes sintomas após a administração, contacte o médico.
Em caso de choque, seguirão as recomendações vigentes para o tratamento do mesmo.
Segurança viral
Quando se administram medicamentos derivados de sangue ou plasma humanos, há que levar a cabo certas medidas para evitar que as infecções passem aos doentes. Tais medidas incluem:
- Uma cuidadosa seleção dos doadores, para excluir aqueles que estão em risco de ser portadores de doenças infecciosas,
- Análise de marcadores específicos de infecções nas doações individuais e nas misturas de plasma,
- A inclusão de etapas no processo de fabricação para eliminar/inativar vírus.
Apesar disto, quando se administram medicamentos derivados da sangue ou plasma humanos, a possibilidade de transmissão de agentes infecciosos não pode ser excluída totalmente. Isto também se refere a vírus emergentes ou de natureza desconhecida ou outros tipos de infecções.
As medidas tomadas são consideradas eficazes para vírus envoltos tais como o vírus da imunodeficiência humana (VIH), o vírus da hepatite B (VHB), o vírus da hepatite C (VHC), e para os vírus não envoltos tais como a hepatite A (VHA) e o parvovirus B19.
O seu médico pode recomendar-lhe que considere a vacinação contra hepatite A e B se receber regularmente produtos com antitrombina derivada de plasma humano.
É altamente recomendável que cada vez que se administre Kybernin P a um doente se deixe constância do nome do medicamento e nº de lote administrado a fim de manter uma relação entre o doente e o lote de produto.
Monitorização clínica e biológica em caso de administração conjunta de antitrombina e heparina:
- A fim de ajustar a dose de heparina e evitar uma excessiva hipocoagulabilidade, devem realizar-se regularmente controles do alcance da anticoagulação (APPT, e quando proceda atividade anti-FXa), a intervalos curtos e em especial nos primeiros minutos/horas posteriores ao início da administração da antitrombina.
- Determinação diária dos níveis de antitrombina, a fim de ajustar a dose individual, devido ao risco de diminuição dos níveis de antitrombina como consequência de um tratamento prolongado com heparina não fraccionada.
Uso de Kybernin P com outros medicamentos
Heparina: a reposição de antitrombina durante a administração de heparina em doses terapêuticas aumenta o risco de sangramento. O efeito da antitrombina é potenciado em grande medida pela heparina. A meia-vida da antitrombina pode diminuir consideravelmente pelo tratamento concomitante com heparina, devido à mobilização acelerada da antitrombina. Por conseguinte, a administração simultânea de heparina e antitrombina a um doente com risco elevado de sangramento deve ser monitorizada clinicamente e biologicamente.
Informe o seu médico ou farmacêutico se está utilizando, utilizou recentemente ou pudesse ter que utilizar qualquer outro medicamento.
Gravidez, lactação e fertilidade
A experiência em relação à segurança dos produtos de antitrombina humana para uso em gravidez humana é limitada.
A segurança do uso de Kybernin P na gravidez humana não foi estabelecida em ensaios clínicos controlados. Os estudos em animais de experimentação são insuficientes para avaliar a segurança em relação à reprodução, desenvolvimento do embrião ou do feto, o curso da gestação e o desenvolvimento peri e pós-natal.
Não há experiências negativas em relação ao tratamento durante a gravidez e a lactação.
Por conseguinte, Kybernin P deve ser administrado a mulheres grávidas ou lactantes com défice de antitrombina apenas se estiver claramente indicado, tendo em conta que a gravidez confere um aumento do risco de episódios tromboembólicos nestas doentes.
Se está grávida ou em período de lactação, acredita que possa estar grávida ou tem intenção de engravidar, consulte o seu médico ou farmacêutico antes de utilizar este medicamento. O seu médico avaliará o possível risco para o feto e informá-lo-á se o tratamento com este medicamento é adequado. O seu médico apenas recomendará este tratamento se estiver claramente indicado.
Condução e uso de máquinas
Não existe nenhum indício de que Kybernin P possa afetar a capacidade para conduzir veículos ou manejar maquinaria.
Kybernin P 500 UI contém sódio
Os doentes com dietas pobres em sódio devem ter em conta que Kybernin P 500 UI contém até 44,76 mg (1,947 mmol) de sódio por 500 UI.
3. Como usar Kybernin P
Kybernin P é um medicamento de uso hospitalar, por isso será administrado num hospital pelo pessoal sanitário correspondente.
Kybernin P é administrado preparando uma solução previa, a qual é injectada ou perfundida por via intravenosa lentamente (máximo 4 ml/min).
Siga exactamente as instruções de administração deste medicamento indicadas pelo seu médico ou farmacêutico. Em caso de dúvida, consulte de novo o seu médico ou farmacêutico.
O seu médico indicará com que frequência e a que intervalos deve ser administrado Kybernin P.
O seu médico indicará a duração do seu tratamento com Kybernin P.
Se usar mais Kybernin P do que deve:
Não foram notificados sintomas de sobredosagem com antitrombina.
Em caso de sobredosagem ou administração accidental, consulte o Serviço de Informação Toxicológica. Telefone 91 562 04 20.
Se esquecer de usar Kybernin P:
- Consulte imediatamente o seu médico ou farmacêutico.
- Não administre uma dose dupla para compensar as doses esquecidas.
4. Posíveis efeitos adversos
Assim como todos os medicamentos, este medicamento pode produzir efeitos adversos, embora não todas as pessoas os sofram.
As seguintes reações adversas baseiam-se na experiência pós-comercialização. Nos casos em que se dispõe de dados foram utilizadas as seguintes categorias estándar de frequência:
Muito frequente >1/10
Frequente >1/100 a <1>
Pouco frequentes ≥ 1/1.000 a <1>
Raras ≥ 1/10.000 a <1>
Muito raras <1>
Classificação por Órgãos e Sistemas | Término Preferido | Frequência |
Trastornos do sistema imunológico | Hipersensibilidade/reações anafilácticas, incluindo anafilaxia grave e choque. | Rara |
Trastornos gerais e no local de administração | Pirexia | Rara |
Para informação sobre segurança viral, ver “Advertências e precauções” na secção 2 deste prospecto.
Comunicação de efeitos adversos
Se experimentar qualquer tipo de efeito adverso, consulte o seu médico ou farmacêutico, mesmo que se trate de efeitos adversos que não aparecem neste prospecto. Também pode comunicá-los directamente através do Sistema Espanhol de Farmacovigilância de Medicamentos de Uso Humano: https://www.notificaram.es. Mediante a comunicação de efeitos adversos, pode contribuir para proporcionar mais informação sobre a segurança deste medicamento.
5. Conservação de Kybernin P
Mantenha este medicamento fora da vista e do alcance das crianças.
Não conserve a temperatura superior a 25 ºC. Não congele.
Não utilize este medicamento após a data de caducidade que aparece no envase após EXP. A data de caducidade é o último dia do mês que se indica.
Não use soluções que estejam turvas ou apresentem resíduos (depósitos/partículas).
Após a reconstituição, a estabilidade físico-química foi demonstrada para um tempo de 8 horas a temperatura ambiente (máx. 25 ºC). Desde um ponto de vista microbiológico e dado que Kybernin P não contém conservantes, a solução reconstituída deve ser utilizada imediatamente. Se isso não for possível, não armazene mais de 8 horas a temperatura ambiente (máximo 25 ºC).
A eliminação do medicamento não utilizado ou do material de desecho será realizada de acordo com as normativas locais.
Os medicamentos não devem ser jogados nos desgotos nem na lixeira. Pergunte ao seu farmacêutico como se livrar dos envases e dos medicamentos que já não precisa. Desta forma, ajudará a proteger o meio ambiente.
6. Conteúdo do envase e informação adicional
Composição de Kybernin P 500 UI
- O princípio ativo é antitrombina III. Cada frasco liofilizado contém 500 UI de antitrombina III. A solução reconstituída contém aproximadamente 50 UI de antitrombina III/ml de antitrombina derivada de plasma humano quando reconstituída com 10 ml de água para preparações injetáveis.
A potência (UI) é determinada utilizando o método do Substrato Cromogênico de acordo com a Farmacopeia Europeia. A atividade específica de Kybernin P é aproximadamente 5,3 UI/mg de proteína.
- Os demais componentes são: glicina, cloreto de sódio, citrato de sódio, ácido clorídrico ou hidróxido de sódio (para ajustar o pH) e água para preparações injetáveis.
Ver seção 2 para informação importante sobre alguns dos excipientes.
Aspecto do produto e conteúdo do envase
Pó e dissolvente para solução injetável e para perfusão.
O envase de comercialização contém um frasco de injeção de vidro moldado tipo II (segundo a Farm. Eur.), incolor e selado com um tampão de borracha, disco de plástico e cápsula de alumínio contendo o liofilizado, um frasco com 10 ml de água para preparações injetáveis (dissolvente para a preparação da solução) e um trasvasador.
Apresentações:
Envase individual de Kybernin P 500 UI:
1 frasco de liofilizado
1 frasco com 10 ml de água para preparações injetáveis
1 trasvasador
Envase clínico de Kybernin P 500 UI:
10 frascos de liofilizado
10 frascos com 10 ml de água para preparações injetáveis
10 trasvasadores
Pode ser que apenas alguns tamanhos de envases sejam comercializados.
Titular da autorização de comercialização e responsável pela fabricação
Titular da autorização de comercialização
CSL Behring, S.A.
Rua Tarragona 157, planta 18
08014 Barcelona - Espanha
Responsável pela fabricação
CSL Behring GmbH
Emil-von-Behring-Str. 76
35041 Marburg - Alemanha
Data da última revisão deste prospecto:Novembro 2020
A informação detalhada e atualizada deste medicamento está disponível na página web da Agência Espanhola de Medicamentos e Produtos Sanitários (AEMPS) http://www.aemps.gob.es
Esta informação está destinada unicamente a profissionais do setor sanitário:
Dosagem
No déficit congênito, a dose deve ser individualizada para cada paciente, tendo em conta a história familiar no que respeita a episódios tromboembólicos, os fatores de risco clínico do paciente e as provas de laboratório.
A dosagem e duração da terapia de substituição no déficit adquirido dependem do nível de antitrombina plasmática, da presença de sinais de mobilização aumentada, do transtorno subjacente e da gravidade do quadro clínico do paciente. A dose e a frequência de administração devem basear-se sempre na eficácia clínica e nas provas de laboratório em cada caso em particular.
O número de unidades de antitrombina administradas expressa-se em Unidades Internacionais (UI), em relação ao padrão da Organização Mundial da Saúde (OMS) vigente para a antitrombina. A atividade plasmática de antitrombina expressa-se como um percentual (em relação ao plasma humano normal) ou em Unidades Internacionais (em relação a um padrão internacional para antitrombina em plasma).
Uma unidade internacional (UI) de atividade de antitrombina é equivalente à quantidade de antitrombina em 1 ml de plasma humano normal. O cálculo da dose necessária de antitrombina baseia-se no achado empírico de que 1 Unidade Internacional (UI) de antitrombina por kg de peso corporal eleva a atividade de antitrombina plasmática em aproximadamente 1,5%.
A dose inicial é determinada mediante a fórmula seguinte:
Unidades requeridas = peso corporal [kg] x (100 – atividade atual da antitrombina [%]) x 2/3.
A atividade de antitrombina que deve ser alcançada inicialmente depende do estado clínico. Quando se estabelece que a substituição com antitrombina está indicada, a dose deve ser suficiente para alcançar a atividade de antitrombina desejada e para manter um nível eficaz. A dose deve ser determinada e monitorizada de acordo com as provas de laboratório da atividade de antitrombina, que se realizarão, pelo menos, duas vezes ao dia até a estabilização do paciente, posteriormente uma vez ao dia, preferentemente imediatamente antes da seguinte perfusão. O ajuste da dose deve ter em conta tanto os sinais de produção aumentada de antitrombina de acordo com as provas de laboratório como a evolução clínica. A atividade de antitrombina deve manter-se acima de 80% durante o tratamento, a não ser que o estado clínico indique um nível de eficácia diferente.
A dose inicial habitual na deficiência congênita é de 30-50 UI/kg.
Portanto, a dose e a frequência de administração, assim como a duração do tratamento devem ajustar-se aos dados biológicos e situação clínica.
População pediátrica:
Kybernin P não é recomendado para uso em crianças menores de 6 anos devido à escassez de dados.
Com base na experiência clínica, não pode recomendar-se o uso de antitrombina no tratamento do SDRI (Síndrome de distresse respiratório infantil) em crianças prematuras.
Instruções para a correta administração do preparado
Instruções gerais
O pó liofilizado deve ser reconstituído completamente, sob condições assépticas, com o dissolvente acompanhante. Obtem-se uma solução transparente ou ligeiramente opalescente.
O diluente apropriado é uma solução de albumina humana a 5%. Para preparar diluições de uma titulação de até 1:5, podem ser utilizadas também: solução Ringer lactato, solução salina fisiológica, solução de glicose a 5% ou poligelina.
O uso de amidos hidroxietilados não é recomendado como dissolvente (para perfusão), pois se observou uma perda de atividade de antitrombina.
Este medicamento não deve ser misturado com outros medicamentos na seringa/equipamento de perfusão. Dopamina, dobutamina e furosemida não devem ser administradas pelo mesmo acesso venoso.
O produto deve ser administrado por via intravenosa. Velocidade de perfusão máxima: 4 ml/min.
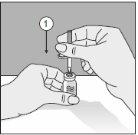
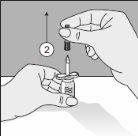
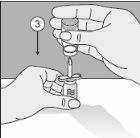
Reconstituição
Para um manuseio correto do Transofix® de dupla ponta, siga os passos a seguir:
|
|
|
|
|
|
|
|
|
|
|
|
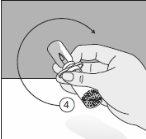
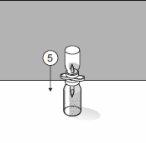
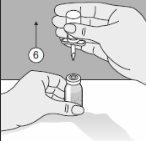
- País de registo
- Substância ativa
- Requer receita médicaSim
- Fabricante
- Esta informação é apenas para referência e não constitui aconselhamento médico. Consulte sempre um médico antes de tomar qualquer medicamento. A Oladoctor não se responsabiliza por decisões médicas baseadas neste conteúdo.
- Alternativas a KYBERNIN P 500 UI Pó e Solvente para Solução Injectável ou PerfusãoForma farmacêutica: PERFURAÇÃO INJETÁVEL, 1.000 UI Antitrombina III HumanaSubstância ativa: antithrombin IIIFabricante: Octapharma S.A.Requer receita médicaForma farmacêutica: PERFURAÇÃO INJETÁVEL, 500 UI antitrombina III humanaSubstância ativa: antithrombin IIIFabricante: Octapharma S.A.Requer receita médicaForma farmacêutica: INJETÁVEL, 1000 UI antitrombinaSubstância ativa: antithrombin IIIFabricante: Csl Behring S.A.Requer receita médica
Alternativas a KYBERNIN P 500 UI Pó e Solvente para Solução Injectável ou Perfusão noutros países
As melhores alternativas com o mesmo princípio ativo e efeito terapêutico.
Alternativa a KYBERNIN P 500 UI Pó e Solvente para Solução Injectável ou Perfusão em Polónia
Alternativa a KYBERNIN P 500 UI Pó e Solvente para Solução Injectável ou Perfusão em Ukraine
Médicos online para KYBERNIN P 500 UI Pó e Solvente para Solução Injectável ou Perfusão
Avaliação de posologia, efeitos secundários, interações, contraindicações e renovação da receita de KYBERNIN P 500 UI Pó e Solvente para Solução Injectável ou Perfusão – sujeita a avaliação médica e regras locais.









Receba novidades da plataforma e promoções exclusivas
Fique a par das atualizações da Oladoctor e receba promoções exclusivas para subscritores.

