

COFACT 500 UI PÓ E SOLVENTE PARA SOLUÇÃO INJETÁVEL
Pergunte a um médico sobre a prescrição de COFACT 500 UI PÓ E SOLVENTE PARA SOLUÇÃO INJETÁVEL



Como usar COFACT 500 UI PÓ E SOLVENTE PARA SOLUÇÃO INJETÁVEL
Introdução
Prospecto: informação para o utilizador
Cofact 500UI pó e dissolvente para solução injetável.
Complexo de protrombina humana
Leia todo o prospecto atentamente antes de começar a usar este medicamento, porque contém informações importantes para si.
- Conserva este prospecto, porque pode ter que voltar a lê-lo.
- Se tiver alguma dúvida, consulte o seu médico ou farmacêutico.
- Se experimentar efeitos adversos, consulte o seu médico ou farmacêutico, mesmo que se trate de efeitos adversos que não aparecem neste prospecto. Ver secção 4.
Conteúdo do prospecto
- O que é Cofact e para que é utilizado
- O que precisa saber antes de começar a usar Cofact
- Como usar Cofact
- Possíveis efeitos adversos
- Conservação de Cofact
- Conteúdo do envase e informação adicional
1. O que é Cofact e para que é utilizado
Cofact contém como princípios ativos, os factores II, VII, IX e X, que são factores de coagulação do sangue humano.
Estes factores são componentes naturais do sangue humano, e são comumente chamados de complexo de protrombina. São dependentes da vitamina K. Se houver uma deficiência de um ou mais destes factores, podem ocorrer distúrbios da coagulação sanguínea. Como resultado disto, há uma maior tendência ao sangramento. A administração de Cofact ajuda a complementar esta deficiência, combatendo ou prevenindo hemorragias.
Cofact pode ser utilizado para:
O tratamento ou prevenção de sangramentos, devidos a
- deficiência adquirida dos factores de coagulação do complexo de protrombina. Por exemplo, em caso de deficiência causada pelo tratamento com antagonistas da vitamina K ou em caso de sobredosagem de antagonistas da vitamina K, quando se requer uma rápida correção desta deficiência.
- deficiências congénitas de um dos factores de coagulação dependentes da vitamina K, quando não se dispõe de produtos purificados do factor de coagulação específico.
2. O que precisa saber antes de começar a usar Cofact
Não use Cofact
- se é alérgico (hipersensível) a algum dos princípios ativos ou a algum dos outros componentes deste medicamento (incluídos na secção 6).
Advertências e precauções
Consulte o seu médico especializado em distúrbios da coagulação antes de receber este medicamento.
- Se tem uma deficiência adquirida dos factores de coagulação dependentes da vitamina K (por exemplo, causada pelo tratamento com medicamentos antagonistas da vitamina K), Cofact só deve ser utilizado quando é necessária uma rápida correção dos níveis do complexo de protrombina, como no caso de um sangramento grave ou uma intervenção cirúrgica de urgência. Em outros casos, geralmente é suficiente a redução da dose do medicamento antagonista da vitamina K e/ou a administração de vitamina K.
- Se recebe um medicamento antagonista da vitamina K, pode apresentar um maior risco de formação de coágulos. Neste caso, o tratamento com Cofact pode potenciar este risco.
- Se nasceu com uma deficiência de algum dos factores de coagulação dependentes da vitamina K (deficiência congénita), devem ser utilizados produtos específicos para o factor de coagulação quando estiverem disponíveis.
- Se ocorre uma reação alérgica ou de tipo anafiláctico, deve interromper-se a perfusão com Cofact imediatamente.
Existe um risco de trombose ou coagulação intravascular disseminada (ou seja, formação de coágulos de sangue nos vasos sanguíneos) quando se recebe este medicamento, especialmente quando se recebe repetidamente.
- Seu médico comprovará se a administração deste medicamento representa um risco de trombose (ver secção 4). As seguintes pessoas têm uma maior probabilidade de sofrer uma trombose:
- pessoas que sofreram um infarto do miocárdio ou que tiveram (ou ainda têm) outras doenças das artérias do coração
- pessoas com doenças do fígado
- recém-nascidos (neonatos)
- pessoas que se vão submeter em breve a uma operação ou que se operaram recentemente
- pessoas com maior probabilidade de sofrer complicações de coagulação (por exemplo, antecedentes de acontecimentos tromboembólicos ou coagulação intravascular disseminada)
Seu médico considerará cuidadosamente o benefício do tratamento com este medicamento em comparação com o risco destas complicações.
Segurança viral
Quando os medicamentos são preparados a partir de sangue ou plasma humanos, são adotadas certas medidas para prevenir a transmissão de infecções aos pacientes. Estas medidas incluem:
- uma cuidadosa seleção dos doadores de sangue e plasma para assegurar que se exclua as pessoas com risco de ser portadoras de infecções,
- a análise de cada doação e das misturas de plasma para detectar a presença de sinais de vírus/infecções,
- a inclusão de etapas no processamento de sangue ou plasma que podem inativar ou eliminar os vírus.
Apesar destas medidas, quando se administram medicamentos preparados a partir de sangue ou plasma humanos, não se pode excluir totally a possibilidade de transmissão de infecções. Isso também se refere a qualquer vírus emergente ou de natureza desconhecida e a outros tipos de infecções.
As medidas adotadas são consideradas eficazes para vírus envoltos como o vírus da imunodeficiência humana (VIH), o vírus da hepatite B (VHB) e o vírus da hepatite C (VHC) e para o vírus não envolto da hepatite A (VHA). As medidas adotadas podem ter um valor limitado contra outros vírus não envoltos como o parvovirus B19. A infecção por parvovirus B19 pode ser grave em mulheres grávidas (infecção do feto) e em pessoas cujo sistema imunológico está deprimido ou que têm algum tipo de anemia (p. ex., doença falciforme ou anemia hemolítica).
Recomenda-se encarecidamente que cada vez que se lhe administre uma dose deste medicamento, se registe o nome e número de lote do produto, com o fim de ter um historial dos lotes utilizados.
Crianças e adolescentes
Não se dispõe de dados em relação ao uso deste medicamento em crianças ou adolescentes.
Outros medicamentos e Cofact
Informa ao seu médico ou farmacêutico se está utilizando, utilizou recentemente ou pudesse ter que utilizar qualquer outro medicamento, mesmo os adquiridos sem receita médica.
Não se dispõe de informação relativa às possíveis interações entre Cofact e outros medicamentos, a exceção dos anticoagulantes.
Gravidez e lactação
Se está grávida ou em período de lactação, acredita que possa estar grávida ou tem intenção de engravidar, consulte o seu médico ou farmacêutico antes de utilizar este medicamento. Se está grávida ou em período de lactação, o seu médico lhe administrará Cofact apenas se estiver claramente indicado.
Condução e uso de máquinas
Não se realizaram estudos sobre os efeitos na capacidade de conduzir ou usar máquinas.
Cofact contém sódio
Cofact contém até 448 mg de sódio (componente principal do sal de mesa ou para cozinhar) por 100 ml. Isso equivale a até 22% da ingestão diária máxima recomendada de sódio para um adulto. Tenha isso em conta se segue uma dieta controlada de sódio.
3. Como usar Cofact
Seu tratamento deve ser iniciado, administrado e monitorizado por um médico com experiência no tratamento dos distúrbios da coagulação. Seu médico determinará a quantidade de Cofact que você precisa para o tratamento ou prevenção de hemorragias devidas ao uso de anticoagulantes ou em caso de deficiência congénita de um dos factores de coagulação dependentes da vitamina K.
A dose exata depende de:
- a gravidade da sua doença
- seu peso
- os factores de coagulação que você precisa
- a quantidade desses factores no seu sangue (seu nível no sangue).
Em caso de deficiência congénita dos factores de coagulação, é importante determinar periodicamente os níveis no sangue dos factores de coagulação.
A informação para profissionais de saúde encontra-se no final do prospecto.
Se usa mais Cofact do que deve
Seu médico deve comprovar regularmente o estado dos seus coágulos durante o tratamento. As doses elevadas de concentrado de complexo de protrombina foram associadas a casos de infarto do miocárdio, coagulação intravascular disseminada e um aumento da formação de coágulos em um vaso sanguíneo em pacientes com risco de sofrer estas complicações.
4. Possíveis efeitos adversos
Como todos os medicamentos, este medicamento pode produzir efeitos adversos, embora nem todas as pessoas os sofram.
Foram observados os seguintes efeitos adversos:
Frequentes(pode afetar menos de 1 em cada 10 pessoas):
- existe um risco de formação de coágulos sanguíneos (ver secção 2)
Pouco frequentes(pode afetar menos de 1 em cada 100 pessoas):
- existe um risco de descida da pressão arterial
A frequência dos seguintes efeitos secundários é desconhecida(a frequência não pode ser estimada a partir dos dados disponíveis):
- Reações de hipersensibilidade ou alérgicas (ver secção 2)
- Ataque ao coração
- Náuseas, vómitos
- Verdade no local de uma perfusão, irritação no local de uma perfusão, inchaço no local de uma perfusão, mal-estar
- Aumento temporário dos resultados dos testes hepáticos
- Acidente vascular cerebral, tonturas
- Embolia pulmonar, dificuldade para respirar
- Sudorese excessiva, picazão na pele, urticária, erupção cutânea
Os pacientes com deficiência de um dos factores de coagulação II, VII, IX ou X podem desenvolver anticorpos contra estes factores como resultado de usar Cofact. Nesse caso, a atividade do produto não será óptima.
Comunicação de efeitos adversos
Se experimenta qualquer tipo de efeito adverso, consulte o seu médico ou farmacêutico, mesmo que se trate de possíveis efeitos adversos que não aparecem neste prospecto. Também pode comunicá-los directamente através do Sistema Espanhol de Farmacovigilância de Medicamentos de Uso Humano: https://www.notificaRAM.es. Mediante a comunicação de efeitos adversos, você pode contribuir para fornecer mais informações sobre a segurança deste medicamento.
5. Conservação de Cofact
Mantenha este medicamento fora da vista e do alcance das crianças.
Não utilize este medicamento após a data de validade que aparece na caixa e na etiqueta do frasco após CAD. A data de validade é o último dia do mês que se indica.
Conservar em frigorífico (2 °C – 8 °C). Não congelar. Conservar o frasco no embalagem exterior para protegê-lo da luz.
Cofact pode ser conservado a ou abaixo de 25 °C durante seis meses. A data em que o medicamento alcançou a temperatura ambiente deve ser anotada no envase. Se não for utilizado durante os seis meses conservado a temperatura ambiente, o produto deve ser descartado.
Uma vez retirado o produto do frigorífico, não deve ser reintroduzido nele.
Foi demonstrada a estabilidade do produto dissolvido até 3 horas a 15 °C - 25 °C. No entanto, para prevenir a contaminação, o produto dissolvido deve ser utilizado imediatamente.
Os medicamentos não devem ser jogados nos esgotos nem na lixeira. Pergunte ao seu farmacêutico como se livrar dos envases e dos medicamentos que já não precisa. Desta forma, ajudará a proteger o meio ambiente.
6. Conteúdo do envase e informação adicional
Composição de Cofact
- Os princípios ativos são os factores de coagulação II, VII, IX e X e outros princípios ativos são as proteínas C e proteína S.
- Um frasco de Cofact 500 UI contém 500 UI de factor IX; 280-700 UI de factor II; 140-400 UI de factor VII e 280-700 UI de factor X; 222-780 UI de proteína C; 20-160 UI de proteína S.
Após dissolver na água para preparações injetáveis que se fornece, a solução para injetáveis pronta para uso contém:
- Não menos de 14 UI e não mais de 35 UI de factor II por ml;
- Não menos de 7 UI e não mais de 20 UI de factor VII por ml;
- 25 UI de factor IX por ml;
- Não menos de 14 UI e não mais de 35 UI de factor X por ml;
- Não menos de 11 UI e não mais de 39 UI de proteína C por ml;
- Não menos de 1 UI e não mais de 8 UI de proteína S por ml.
Os demais componentes são citrato de sódio, cloreto de sódio e antitrombina.
Aspecto do produto e conteúdo do envase
Cofact é fornecido como pó e dissolvente para uma solução injetável.
Cofact pó para injetável é um pó de cor azulada. O dissolvente é um líquido claro, incolor e sem partículas visíveis. A solução preparada e pronta para a injeção é uma solução de cor azulada.
Conteúdo do envase de 500UI
1 frasco de pó de 500 UI
1 frasco de 20 ml de água para preparações injetáveis
1 dispositivo de transferência nextaro v
Pode ser que apenas alguns tamanhos de envases sejam comercializados.
Titular da autorização de comercialização e responsável pela fabricação
Prothya Biosolutions Netherlands B.V.
Plesmanlaan 125
NL-1066 CX Amesterdão
Países Baixos
Este medicamento está autorizado nos estados membros do Espaço Económico Europeu com os seguintes nomes:
Áustria, Bélgica, Finlândia, França, Alemanha, Islândia, Itália, Luxemburgo, Países Baixos e Espanha: Cofact.
Suécia: Thyaplex
Data da última revisão deste prospecto: Agosto 2024
--------------------------------------------------------------------------------------------------------------------
Esta informação está destinada apenas a profissionais do setor sanitário:
Composição qualitativa e quantitativa
Cofact apresenta-se sob a forma de pó e diluente para solução injetável que contém o complexo de protrombina humana. O produto contém nominalmente as seguintes UI dos factores de coagulação humanos:
Cofact 500 UI | Depois da reconstituição*(UI/ml) | ||
Princípios ativos | |||
Fator II de coagulação | 280 – 700 | 14 – 35 | |
Fator VII de coagulação | 140 – 400 | 7 – 20 | |
Fator IX de coagulação | 500 | 25 | |
Fator X de coagulação | 280 – 700 | 14 – 35 | |
Outros princípios ativos | |||
Proteína C | 222 – 780 | 11 – 39 | |
Proteína S | 20 – 160 | 1 – 8 | |
*Depois da reconstituição com 20 ml de água para preparações injetáveis.
O conteúdo total de proteínas por frasco de 500 UI é de 260 – 700 mg. A atividade específica do produto é ≥ 0,6 UI/mg, expressa como atividade do fator IX.
A atividade de todos os factores de coagulação, bem como das proteínas C e S (antígenos) foi determinada de acordo com os padrões atuais da OMS ou da Farmacopeia Europeia.
Após a reconstituição, este medicamento contém 125 – 195 mmol de sódio/l, até 89,6 mg de sódio por frasco de 500 UI.
Posologia e forma de administração
Posologia
A seguir são fornecidas apenas diretrizes gerais de dosagem. O tratamento deve ser iniciado sob a supervisão de um médico com experiência no tratamento de distúrbios de coagulação. A dose e duração do tratamento de substituição dependem da gravidade do distúrbio, da localização e intensidade do sangramento e do quadro clínico do paciente.
A posologia e frequência de administração devem ser calculadas de forma individual para cada paciente. Os intervalos de dosagem devem ser adaptados às diferentes meias-vidas circulantes dos diferentes factores de coagulação no complexo de protrombina. Os requisitos posológicos individuais só podem ser calculados com base na determinação periódica dos níveis plasmáticos individuais dos factores de coagulação em questão, ou da análise global dos níveis do complexo de protrombina (tempo de protrombina, INR) e na monitorização contínua da situação clínica do paciente.
No caso de intervenções cirúrgicas maiores, é imprescindível uma monitorização precisa do tratamento de substituição, por meio de análises de coagulação (avaliações do fator de coagulação específico e/ou análises globais dos níveis de complexo de protrombina).
Sangramento e profilaxia perioperatória de sangramentos durante o tratamento com antagonistas da vitamina K:
A dose dependerá do INR antes do tratamento, do INR desejado e do peso corporal. Nas seguintes tabelas são fornecidas doses aproximadas que são necessárias para a normalização do INR a diferentes níveis iniciais de INR.
As tabelas de dose só representam diretrizes gerais de dosagem, o que não pode substituir a avaliação individual da dose para cada paciente e o controle rigoroso do INR e outros parâmetros de coagulação durante o tratamento.
Doses recomendadas de Cofact em ml para alcançar um INR desejado ≤ 2,1
INR inicial Peso corporal | 7,5 | 5,9 | 4,8 | 4,2 | 3,6 | 3,3 | 3,0 | 2,8 | 2,6 | 2,5 | 2,3 | 2,2 |
50kg | 40 | 40 | 40 | 30 | 30 | 30 | 20 | 20 | X | X | X | X |
60kg | 50 | 50 | 40 | 40 | 30 | 30 | 30 | 20 | X | X | X | X |
70kg | 60 | 50 | 50 | 50 | 40 | 40 | 30 | 30 | X | X | X | X |
80kg | 60 | 60 | 60 | 50 | 50 | 40 | 40 | 30 | X | X | X | X |
90kg | 60 | 60 | 60 | 60 | 50 | 50 | 40 | 30 | X | X | X | X |
100kg | 60 | 60 | 60 | 60 | 60 | 50 | 40 | 40 | X | X | X | X |
Doses recomendadas de Cofact em ml para alcançar um INR desejado ≤ 1,5
INRinicial Peso corporal | 7,5 | 5,9 | 4,8 | 4,2 | 3,6 | 3,3 | 3,0 | 2,8 | 2,6 | 2,5 | 2,3 | 2,2 |
50kg | 60 | 60 | 60 | 50 | 50 | 50 | 40 | 40 | 30 | 30 | 30 | 30 |
60kg | 80 | 70 | 70 | 60 | 60 | 60 | 50 | 50 | 40 | 40 | 40 | 30 |
70kg | 90 | 80 | 80 | 70 | 70 | 70 | 60 | 60 | 50 | 40 | 40 | 40 |
80kg | 100 | 100 | 90 | 90 | 90 | 80 | 80 | 70 | 60 | 50 | 50 | 40 |
90kg | 100 | 100 | 100 | 90 | 90 | 90 | 80 | 80 | 70 | 60 | 50 | 40 |
100kg | 100 | 100 | 100 | 100 | 100 | 90 | 90 | 80 | 70 | 70 | 60 | 50 |
As doses são calculadas com base na concentração de fator IX em Cofact, devido à sua meia-vida relativamente curta e baixo rendimento após a perfusão em comparação com os outros factores de coagulação em Cofact. Pressupõe-se que uma concentração plasmática média de fator IX ≥ 30% é suficiente para alcançar um INR ≥ 2,1 e ≥ 60% para alcançar um INR ≥ 1,5. As quantidades calculadas são arredondadas em múltiplos de 10 ml e foi estabelecido um limite superior de 60 ou 100 ml no total (ver as tabelas acima). Os valores de INR desejados são recomendados pela Federação de Serviços Holandeses de Trombose e são do mesmo nível que as recomendações inglesas e alemãs.
A correção do distúrbio de hemostasia induzido pelos antagonistas da vitamina K persiste durante cerca de 6-8 horas. No entanto, os efeitos da vitamina K, se administrada ao mesmo tempo, normalmente são alcançados em um período de 4-6 horas. Portanto, normalmente não é necessário repetir o tratamento com complexo de protrombina humana quando se administra vitamina K.
Devido a que essas recomendações são empíricas e que a recuperação e a duração do efeito podem variar, é obrigatória a monitorização do INR durante o tratamento.
Sangramento e profilaxia perioperatória da deficiência congênita de qualquer dos factores de coagulação dependentes da vitamina K, quando não se dispõe do produto de fator de coagulação específico:
O cálculo da dose necessária para o tratamento é baseado no dado empírico de que aproximadamente 1 UI de fator VII ou de fator IX por kg de peso corporal aumenta a atividade plasmática do fator VII ou IX, respectivamente, em 0,01 UI/ml; e 1 UI de fator II ou X por kg de peso corporal aumenta a atividade plasmática do fator II ou X em 0,02 e 0,017 UI/ml, respectivamente.
A dose de um fator específico administrado é expressa em Unidades Internacionais (UI), que estão relacionadas com o padrão atual da OMS para cada fator. A atividade plasmática de um fator de coagulação específico é expressa, ou bem como um percentual (relativo ao plasma normal) ou em Unidades Internacionais (relativas ao padrão internacional para o fator de coagulação específico).
Uma Unidade Internacional (UI) de atividade de um fator de coagulação é equivalente à quantidade contida em um ml de plasma humano normal.
Por exemplo, o cálculo da dose necessária de fator X é baseado no dado empírico de que 1 Unidade Internacional (UI) de fator X por kg de peso corporal aumenta a atividade plasmática do fator X em 0,017 UI/ml. A dose necessária é determinada utilizando a seguinte fórmula:
Unidades necessárias = peso corporal (kg) x incremento desejado do fator X (UI/ml) x 60
Onde 60 (ml/kg) é o valor recíproco da recuperação estimada.
Se a recuperação individual for conhecida, este valor deve ser usado para o cálculo.
População pediátrica
Não foi estabelecida a segurança e eficácia do uso de Cofact em pacientes pediátricos.
Forma de administração
Para consultar as instruções de reconstituição do medicamento antes da administração, ver seção 6.6. “Precauções especiais de eliminação e outras manipulações”. Cofact deve ser administrado por via intravenosa.
Recomenda-se administrar o produto reconstituído a uma velocidade de aproximadamente 2 ml por minuto.
Incompatibilidades
Este medicamento não deve ser misturado com outros medicamentos.
Cofact é compatível com material de polipropileno. O tratamento pode falhar devido à adsorção do fator de coagulação na superfície interna de outros equipamentos de injeção/perfusão.
Período de validade
3 anos.
Após a reconstituição, foi demonstrada a estabilidade fisicoquímica em uso durante 3 horas a 15 °C – 25 °C. Desde o ponto de vista microbiológico, o produto deve ser utilizado imediatamente após a reconstituição. Se não for utilizado imediatamente, os tempos de conservação em uso e as condições antes do uso são responsabilidade do usuário.
Precauções especiais de eliminação e outras manipulações
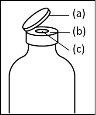
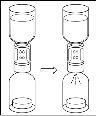
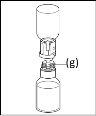
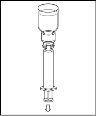
Instruções gerais para o uso de um dispositivo de transferência nextarov
- A fração proteica desidratada deve ser dissolvida com 20 ml de água para preparações injetáveis. Se os frascos fechados de pó e dissolvente (água para preparações injetáveis) forem armazenados a 2 °C - 8 °C, é necessário permitir que atinjam a temperatura ambiente (15 °C - 25 °C) antes de dissolver a preparação. Esta temperatura deve ser mantida durante a reconstituição. Se um banho de água for utilizado para o aquecimento, é necessário ter cuidado para evitar que a água entre em contato com as tampas de borracha ou as tampas abertas do frasco. A temperatura do banho de água não deve ultrapassar 37 °C.
- Durante o procedimento descrito a seguir, deve ser aplicada uma técnica asséptica. Antes de abrir o envase do dispositivo de transferência, certifique-se de remover as tampas abertas dos frascos de pó e dissolvente e desinfetar o bordo do pescoço e as tampas de borracha com uma solução antiséptica e permita que sequem. Não toque as tampas de borracha do frasco de dissolvente nem do frasco de pó.
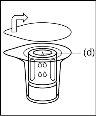
- Como resultado do vácuo no frasco de pó, o dissolvente é transferido automaticamente para o frasco de pó.
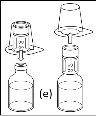
- Como regra geral, o pó deve ser dissolvido completamente em 10 minutos para formar uma solução de cor azul. A solução deve ser transparente ou ligeiramente opalescente. Não utilize soluções turvas ou com depósitos. A solução deve ser inspecionada visualmente para detectar partículas e decoloração antes de sua administração.
- Qualquer produto não utilizado ou material de descarte deve ser eliminado de acordo com a regulamentação local.
- Procedimento para o uso de um dispositivo de transferência nextarov
|
|
|
|
|
|
| |
Nota: o dispositivo de transferência deve ser fixado primeiro ao frasco de dissolvente e depois ao frasco de pó. Caso contrário, ocorrerá uma perda de vácuo e a transferência do dissolvente não poderá ser realizada. |
|
Aguarde até que o dissolvente seja transferido completamente. Continue segurando a unidade completa formada pelo frasco de dissolvente, o dispositivo de transferência e o frasco de pó, e certifique-se de que permaneça sobre uma superfície plana durante todo o processo de transferência. Uma vez transferido o dissolvente, com ambos os frascos ainda unidos, agite suavemente o frasco de pó até que o produto esteja completamente dissolvido. Para evitar a formação de espuma, não agite o frasco. |
|
|
|
|
|
|
|


- País de registo
- Substância ativa
- Requer receita médicaSim
- Fabricante
- Esta informação é apenas para referência e não constitui aconselhamento médico. Consulte sempre um médico antes de tomar qualquer medicamento. A Oladoctor não se responsabiliza por decisões médicas baseadas neste conteúdo.
- Alternativas a COFACT 500 UI PÓ E SOLVENTE PARA SOLUÇÃO INJETÁVELForma farmacêutica: INJETÁVEL, 1000 UI/frascoSubstância ativa: coagulation factor IX, II, VII and X in combinationFabricante: Csl Behring GmbhRequer receita médicaForma farmacêutica: INJETÁVEL, 500 UISubstância ativa: coagulation factor IX, II, VII and X in combinationFabricante: Csl Behring GmbhRequer receita médicaForma farmacêutica: INJETÁVEL, 250 UISubstância ativa: coagulation factor IX, II, VII and X in combinationFabricante: Prothya Biosolutions Netherlands B.V.Requer receita médica
Alternativas a COFACT 500 UI PÓ E SOLVENTE PARA SOLUÇÃO INJETÁVEL noutros países
As melhores alternativas com o mesmo princípio ativo e efeito terapêutico.
Alternativa a COFACT 500 UI PÓ E SOLVENTE PARA SOLUÇÃO INJETÁVEL em Polónia
Alternativa a COFACT 500 UI PÓ E SOLVENTE PARA SOLUÇÃO INJETÁVEL em Ukraine
Médicos online para COFACT 500 UI PÓ E SOLVENTE PARA SOLUÇÃO INJETÁVEL
Avaliação de posologia, efeitos secundários, interações, contraindicações e renovação da receita de COFACT 500 UI PÓ E SOLVENTE PARA SOLUÇÃO INJETÁVEL – sujeita a avaliação médica e regras locais.









Receba novidades da plataforma e promoções exclusivas
Fique a par das atualizações da Oladoctor e receba promoções exclusivas para subscritores.

