


FEIBA 100 U/mL PÓ E SOLVENTE PARA SOLUÇÃO PARA PERFUSÃO
Pergunte a um médico sobre a prescrição de FEIBA 100 U/mL PÓ E SOLVENTE PARA SOLUÇÃO PARA PERFUSÃO



Como usar FEIBA 100 U/mL PÓ E SOLVENTE PARA SOLUÇÃO PARA PERFUSÃO
Introdução
Prospecto: informação para o utilizador
FEIBA 100U/ml pó e dissolvente para solução para perfusão
complexo coagulante antiinibidor
Leia todo o prospecto atentamente antes de começar a usar este medicamento, porque contém informações importantes para si.
- Conserva este prospecto, porque pode ter que voltar a lê-lo.
- Se tiver alguma dúvida, consulte o seu médico ou farmacêutico.
- Este medicamento foi prescrito apenas para si, e não deve dá-lo a outras pessoas, mesmo que tenham os mesmos sintomas que si, porque pode prejudicá-las.
- Se experimentar efeitos adversos, consulte o seu médico ou farmacêutico, mesmo que se trate de efeitos adversos que não aparecem neste prospecto. Ver secção 4.
Conteúdo do prospecto
- O que é FEIBA e para que é utilizado
- O que precisa saber antes de começar a usar FEIBA
- Como usar FEIBA
- Posíveis efeitos adversos
- Conservação de FEIBA
- Conteúdo do envase e informações adicionais
1. O que é FEIBA e para que é utilizado
FEIBA é uma preparação elaborada a partir de plasma humano, que permite a hemostasia, mesmo quando a quantidade dos factores de coagulação específicos está reduzida ou ausente.
FEIBA é utilizado para o tratamento e a profilaxia de hemorragias em pacientes com hemofilia A com inibidor.
FEIBA é utilizado para o tratamento de hemorragias em pacientes com hemofilia B com inibidor.
FEIBA pode ser utilizado para o tratamento e a profilaxia de hemorragias em pacientes não hemofílicos com inibidor adquirido do factor VIII.
Além disso, FEIBA é utilizado para a profilaxia em intervenções cirúrgicas em pacientes com hemofilia A com inibidor.
FEIBA pode ser utilizado em todos os grupos etários.
2. O que precisa saber antes de começar a usar FEIBA
Informa o seu médico se tem alguma alergia conhecida.
Informa o seu médico se está a seguir uma dieta pobre em sódio.
Não use FEIBA
Deve usar FEIBA apenas nas seguintes circunstâncias se, por exemplo, devido a um título muito elevado de inibidores não se espera nenhuma resposta ao tratamento com o concentrado de factor de coagulação apropriado:
- se é alérgico (hipersensível) ao complexo coagulante antiinibidor ou a algum dos outros componentes deste medicamento (incluídos na secção 6).
- se existe uma coagulação intravascular disseminada (CID). (CID = coagulopatia de consumo, uma condição potencialmente mortal em que há uma coagulação excessiva do sangue, com formação pronunciada de coágulos de sangue nos vasos sanguíneos. Isso provoca um consumo dos factores de coagulação em todo o organismo).
- em caso de infarto de miocárdio, trombose e/ou embolia aguda: FEIBA deve ser utilizado apenas nos episódios hemorrágicos que constituam uma ameaça para a vida.
Advertências e precauções
Consulte o seu médico antes de começar a usar FEIBA porque podem produzir-se reações de hipersensibilidade, como no caso de todos os produtos derivados do plasma que se administram por via intravenosa. Para poder reconhecer uma reação alérgica o mais cedo possível, deve conhecer que os primeiros sintomas potenciais de uma reação de hipersensibilidade podem ser:
- eritema (enrubescimento da pele)
- erupção cutânea
- aparição de habões na pele (urticária)
- picar por todo o corpo
- inchação dos lábios e da língua
- dificuldade para respirar/disneia
- opressão no peito
- malestar geral
- tontura
- queda da tensão arterial
Outros sintomas de reações de hipersensibilidade a produtos derivados do plasma incluem letargia e cansaço.
Se nota algum desses sintomas, deve parar a administração imediatamente e contactar com o seu médico de imediato. Os sintomas descritos podem indicar um choque anafiláctico. Os sintomas intensos requerem um tratamento temprano de urgência.
O seu médico só reutilizará FEIBA em pacientes com suspeita de hipersensibilidade ao produto ou a algum dos seus componentes após avaliar detidamente o benefício esperado e o risco da reexposição e/ou de não esperar nenhuma resposta com outro tratamento preventivo ou terapia alternativa.
- Se experimenta mudanças importantes na tensão arterial ou na frequência do pulso, dificuldades para respirar, tosse ou dor no peito, deve parar a administração imediatamente e contactar com o seu médico. O seu médico iniciará as medidas de diagnóstico e terapêuticas apropriadas.
- Em pacientes com hemofilia com inibidores ou com inibidores adquiridos aos factores de coagulação. Durante o tratamento com FEIBA, estes pacientes podem ter um aumento na tendência a desenvolver hemorragia e um aumento do risco de trombose ao mesmo tempo.
Durante o tratamento com FEIBA, apareceram acontecimentos trombóticos e tromboembólicos, incluindo coagulação intravascular disseminada (CID), trombose venosa, embolia pulmonar, infarto de miocárdio e acidente vascular cerebral. É provável que o uso concomitante com factor VIIa recombinante aumente o risco de desenvolver um acontecimento tromboembólico. Alguns dos acontecimentos tromboembólicos ocorreram com o tratamento de altas doses de FEIBA.
Em um ensaio realizado por outra empresa para avaliar emicizumab (um medicamento para prevenir sangrados em pacientes com hemofilia A), alguns pacientes que sofreram sangrados intercorrentes foram tratados com FEIBA para controlar os sangrados e, alguns desses pacientes, desenvolveram microangiopatia trombótica (MAT). MAT é uma condição grave e potencialmente ameaçadora para a vida. Quando se tem esta condição, pode danificar a parede vascular e podem desenvolver-se coágulos nos vasos sanguíneos pequenos. Em alguns casos, isso pode ocasionar dano nos rins e outros órgãos. Em caso de sangrados intercorrentes enquanto se está em profilaxia com emicizumab, contacte imediatamente com o seu hematologista ou com o seu Centro de Tratamento de Hemofilia.
Quando se administram medicamentos derivados de plasma ou sangue humano, há que levar a cabo certas medidas para evitar que as infecções passem aos pacientes. Tais medidas incluem uma cuidadosa seleção dos doadores, para excluir aqueles que estão em risco de ser portadores de doenças infecciosas, análise de marcadores específicos de infecções nas doações individuais e nas misturas de plasma, assim como a inclusão de etapas no processo de fabricação para eliminar/inativar vírus. Apesar disso, quando se administram medicamentos derivados do sangue ou plasma humanos, a possibilidade de transmissão de agentes infecciosos não se pode excluir totalmente. Isso também se refere a vírus emergentes ou de natureza desconhecida ou outros tipos de infecções.
Estas medidas são consideradas eficazes para vírus envoltos como o vírus da imunodeficiência humana (VIH), vírus da hepatite B e vírus da hepatite C e para os vírus não envoltos da hepatite A. As medidas adotadas podem ter um valor limitado para vírus não envoltos como o parvovirus B19. A infecção por parvovirus B19 pode ser grave para uma mulher grávida (infecção fetal) e para indivíduos cujo sistema imunológico está deprimido ou para pacientes com algum tipo de anemia (por exemplo, doença falciforme ou anemia hemolítica).
É possível que o seu médico lhe recomende vacinar-se contra hepatite A e hepatite B, se a si lhe for administrado de forma regular ou repetida produtos derivados do plasma para os inibidores do factor VIII.
Depois da administração de doses elevadas de FEIBA, o aumento transitório dos anticorpos de superfície da hepatite B transferidos passivamente pode provocar uma interpretação errónea dos resultados positivos do teste serológico.
FEIBA é um derivado de plasma e pode conter substâncias que reagem quando se perfundem aos pacientes, causando a presença de isohemaglutininas (anticorpos que provocam a adesão dos glóbulos vermelhos de outra pessoa). Este processo pode levar a malinterpretar os resultados dos análises de sangue.
Recomenda-se encarecidamente que cada vez que se administre uma dose de FEIBA, se deixe constância do nome do medicamento e número de lote administrado com o fim de manter um registo dos lotes utilizados.
Crianças
A experiência em crianças menores de 6 anos é limitada; deve adaptar-se o mesmo regime posológico que nos adultos para o estado clínico da criança.
Outros medicamentos e FEIBA
Informa o seu médico ou farmacêutico se está a utilizar, utilizou recentemente ou pudesse ter que utilizar qualquer outro medicamento.
Não se realizaram estudos adequados e bem controlados do uso combinado ou sequencial de FEIBA e factor VIIa recombinante, antifibrinolíticos ou emicizumab. Quando se usam antifibrinolíticos sistémicos, como ácido tranexâmico e ácido aminocaproico, durante o tratamento com FEIBA, deve considerar-se a possibilidade de aparecimento de acontecimentos tromboembólicos. Por isso, não se devem utilizar antifibrinolíticos até aproximadamente 6 a 12 horas após a administração de FEIBA.
De acordo com os dados in vitro disponíveis e as observações clínicas, não se pode excluir uma interação potencial medicamentosa com o uso concomitante com factor VIIa recombinante que potencialmente produz um acontecimento tromboembólico. Informa o seu médico se vai a ser tratado com FEIBA após ter recebido emicizumab (um medicamento para prevenir os sangrados em pacientes com hemofilia A) já que há que ter em conta algumas advertências e precauções específicas. O seu médico necessitará fazer-lhe um seguimento estreito.
Como com todos os produtos utilizados para a coagulação sanguínea, FEIBA não deve misturar-se com outros medicamentos antes da sua administração, porque isso pode prejudicar a eficácia e tolerância do produto. É conveniente lavar a via venosa com solução salina isotónica antes e após a administração de FEIBA.
Gravidez, lactação e fertilidade
Se está grávida ou em período de lactação, acredita que possa estar grávida ou tem intenção de engravidar, consulte o seu médico ou farmacêutico antes de utilizar este medicamento.
O seu médico decidirá se FEIBA pode ser usado durante a gravidez e a lactação. Devido ao aumento do risco de trombose durante a gravidez, FEIBA só deve ser administrado sob uma estreita supervisão médica e só se encontrar claramente indicado. Para informação sobre o risco de infecção por parvovirus B19, ver secção advertências e precauções.
Condução e uso de máquinas
Não há nenhum sinal de que FEIBA possa afetar a capacidade para conduzir e utilizar máquinas.
FEIBA contém sódio
500 U
Este medicamento contém aproximadamente 40 mg de sódio (componente principal do sal de mesa/para cozinhar) em cada frasco. Isso equivale a 2% da ingestão diária máxima de sódio recomendada para um adulto.
1 000 U
Este medicamento contém aproximadamente 80 mg de sódio (componente principal do sal de mesa/para cozinhar) em cada frasco. Isso equivale a 4% da ingestão diária máxima de sódio recomendada para um adulto.
2 500 U
Este medicamento contém aproximadamente 200 mg de sódio (componente principal do sal de mesa/para cozinhar) em cada frasco. Isso equivale a 10% da ingestão diária máxima de sódio recomendada para um adulto.
3. Como usar FEIBA
Reconstituir o pó liofilizado de FEIBA com o dissolvente incluído e administre a solução por via intravenosa.
Siga exatamente as instruções de administração deste medicamento indicadas pelo seu médico. Em caso de dúvida, consulte de novo o seu médico ou farmacêutico.
O seu médico determinará a frequência e a dose requerida para si pessoalmente, tendo em conta a gravidade do distúrbio da coagulação sanguínea, a localização e a magnitude da hemorragia, e o estado clínico e a resposta à preparação. Não mude a dosagem estabelecida pelo seu médico e não suspenda a administração do preparado.
Se tem a impressão de que o efeito de FEIBA é demasiado forte ou demasiado débil, consulte o seu médico ou farmacêutico.
Aquecer o produto a temperatura ambiente ou a temperatura corporal antes da sua administração, se necessário.
FEIBA deve reconstituir-se imediatamente antes da sua administração. A solução deve utilizar-se de imediato (já que a preparação não contém conservantes).
Agitar suavemente até que todo o produto esteja dissolvido. Asegurar que FEIBA esteja completamente dissolvido, porque, de outra forma, passarão menos unidades de FEIBA através do filtro do equipamento.
As soluções de aspecto turvo ou que contenham depósitos devem ser eliminadas adequadamente.
Não reutilizar os envases abertos.
Utilizar apenas a água para preparações injetáveis e o equipamento para a reconstituição incluídos no envase.
Se se utilizam outros equipamentos distintos dos incluídos, asegure-se de usar um filtro adequado de, pelo menos, 149 micrómetros de tamanho de poro.
Não utilizar o produto se o seu sistema de barreira de esterilidade ou o seu envase estão danificados ou mostram qualquer sinal de deterioração.
Não refrigerar após a reconstituição.
Após a completa reconstituição de FEIBA, a injeção ou perfusão deve começar imediatamente e deve completar-se no prazo de 3 horas desde a reconstituição.
A eliminação do medicamento não utilizado e de todos os materiais que tenham estado em contacto com ele será realizada de acordo com a normativa local.
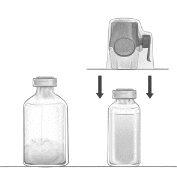
Reconstituição do pó para a preparação de uma solução para perfusão com o equipamento BAXJECT II Hi-Flow:
- Aquecer o frasco de dissolvente sem abrir (água para preparações injetáveis) a temperatura ambiente ou máximo 37°C, se necessário, por exemplo, utilizando um banho de água durante vários minutos.
- Retirar os tampões protectores do frasco de pó e do frasco de dissolvente, e desinfectar os tampões de borracha de ambos os frascos. Colocar os frascos em uma superfície lisa.
- Abrir o envoltório do equipamento BAXJECT II Hi-Flow retirando a lâmina protectora sem tocar o conteúdo do envase (Figura a). Não sacar o equipamento do envoltório.
- Dar a volta ao envoltório e inserir a ponta de plástico transparente através do tampão de borracha do frasco de dissolvente (Figura b). Sacar agora o equipamento BAXJECT II Hi-Flow do seu envoltório (Figura c). Não retire o tampão protector azul do equipamento BAXJECT II Hi-Flow.
- Com o equipamento BAXJECT II Hi-Flow unido ao frasco de dissolvente, inverta o sistema de tal forma que o frasco de dissolvente esteja na parte superior do equipamento. Inserir a ponta de plástico de cor púrpura do equipamento BAXJECT II Hi-Flow através do tampão do frasco de FEIBA. O vácuo fará com que o dissolvente penetre no frasco de FEIBA (Figura d).
- Agitar suavemente, sem sacudir, todo o sistema até que todo o pó esteja dissolvido. Asegure-se de que FEIBA esteja completamente dissolvido, de outra forma o material ativo pode ficar retido no filtro do equipamento.
Figura a | Figura b | Figura c |
|
|
|


Perfusão
Usar uma técnica asséptica durante todo o procedimento!
- Retirar o tampão protector azul do equipamento BAXJECT II Hi-Flow. Conectar firmemente a seringa ao equipamento BAXJECT II Hi-Flow. NÃO INTRODUZIR AR NA SERINGA. (Figura e). Recomenda-se encarecidamente utilizar uma seringa Luer Lock com o objetivo de assegurar uma conexão firme entre a seringa e o equipamento BAXJECT II Hi-Flow (girar a seringa no sentido dos ponteiros do relógio até que se pare quando chegar ao topo).
- Inverter o sistema para que o produto dissolvido se encontre na parte de cima. Introduzir o produto dissolvido na seringa, puxando o êmbolo para trás LENTAMENTE e assegurar que a conexão firme entre o dispositivo BAXJECT II Hi-Flow e a seringa se mantém durante todo o processo enquanto se puxa o êmbolo da seringa (Figura f).
- Desconectar a seringa.
- Se se produzir espuma dentro da seringa, esperar a que a espuma se compacte. Administrar lentamente por via intravenosa a solução com o equipamento de perfusão fornecido.
Figura d | Figura e | Figura f |
|
|
|

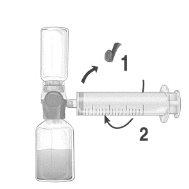

Não exceder uma velocidade de perfusão de 10U de FEIBA/kg por minuto.
Se usa mais FEIBA do que deve
Informa imediatamente o seu médico. A sobredosagem de FEIBA pode aumentar o risco de efeitos adversos, como tromboembolia (formação de um coágulo de sangue com enrubescimento nos vasos sanguíneos), coagulação intravascular disseminada (CID) ou infarto de miocárdio. Alguns dos acontecimentos tromboembólicos notificados produziram-se com doses superiores a 200 U/kg/dia ou com pacientes com outros factores de risco de sofrer acontecimentos tromboembólicos. Se se observam sinais ou sintomas de acontecimento tromboembólico, a perfusão deve ser interrompida imediatamente e devem ser adoptadas as medidas de diagnóstico e terapêuticas necessárias.
Em caso de sobredosagem ou ingestão acidental, consulte imediatamente o seu médico ou farmacêutico ou ligue para o Serviço de Informação Toxicológica, telefone 91 562 04 20, indicando o medicamento e a quantidade ingerida.
4. Efeitos adversos possíveis
Tal como todos os medicamentos, este medicamento pode produzir efeitos adversos, embora nem todas as pessoas os sofram.
Efeitos adversos frequentes(podem afetar até 1 de cada 10 doentes)
Hipersensibilidade, dor de cabeça, tontura, hipotensão, erupção cutânea, anticorpos de superfície da hepatite B positivos.
Efeitos adversos com frequência desconhecida(não pode ser estimada a partir dos dados disponíveis)
Perturbações do sangue e do sistema linfático:coagulação intravascular disseminada (CID), aumento do título de inibidor.
Perturbações do sistema imunológico:reações anafilácticas, erupção cutânea por todo o corpo (urticária).
Perturbações do sistema nervoso:sensação de entorpecimento das extremidades (hipoestesia), sensibilidade anormal ou reduzida (parestesia), acidente cerebrovascular (acidente trombótico, acidente embólico), sonolência, sensação do gosto alterada (disgeusia).
Perturbações cardíacas:ataque cardíaco (infarto do miocárdio), palpitação do coração (taquicardia).
Perturbações vasculares:formação de coágulos de sangue com enrubescimento dos vasos sanguíneos (acontecimentos tromboembólicos, trombose venosa e arterial), aumento da tensão arterial (hipertensão), rubor.
Perturbações respiratórias, torácicas e mediastínicas:obstrução da artéria pulmonar (embolia pulmonar), obstrução do passo do ar (broncoespasmo), pitidos no peito, tosse, dificuldade ao respirar (dispnéia).
Perturbações gastrointestinais:vómitos, diarreia, mal-estar abdominal, sensação de doença (náuseas).
Perturbações da pele e do tecido subcutâneo:sensação de entorpecimento na face, inchação da face, da língua e dos lábios (angioedema), erupção cutânea por todo o corpo (urticária), picor (prurito).
Perturbações gerais e alterações no local de administração:dor no local da injeção, mal-estar geral, sensação de calor, arrepios, febre, dor no peito, mal-estar no peito.
Exames complementares:queda da tensão arterial, aumento do nível de dímero D de fibrina no sangue.
A perfusão intravenosa rápida pode causar uma dor pontiaguda e uma sensação de entorpecimento na face e nas extremidades, assim como uma diminuição da tensão arterial.
Foram notificados casos de infarto do miocárdio após a administração de doses superiores à dose máxima diária e/ou com administrações prolongadas e/ou da presença de fatores de risco tromboembólicos.
Comunicação de efeitos adversos
Se experimentar qualquer tipo de efeito adverso, consulte o seu médico, mesmo que se trate de possíveis efeitos adversos que não aparecem neste prospecto. Também pode comunicá-los diretamente através do Sistema Español de Farmacovigilância de Medicamentos de Uso Humano: www.notificaram.es.
Ao comunicar efeitos adversos, você pode contribuir para fornecer mais informações sobre a segurança deste medicamento.
5. Conservação de FEIBA
Mantenha este medicamento fora da vista e do alcance das crianças.
Não conserve a uma temperatura superior a 25 °C. Não congele.
Conservar no embalagem original para protegê-lo da luz.
Não utilize este medicamento após a data de caducidade que aparece na etiqueta e no envase. A data de caducidade é o último dia do mês que se indica.
Os medicamentos não devem ser jogados nos desagueiros nem na lixeira. Pergunte ao seu farmacêutico como se desfazer dos envases e dos medicamentos que já não precisa. Dessa forma, ajudará a proteger o meio ambiente.
6. Conteúdo do envase e informação adicional
Composição de FEIBA
Pó
- O princípio ativo por frasco é complexo coagulante antiinibidor.
- 1 ml contém 100 U de complexo coagulante antiinibidor.
- FEIBA 100 U/ml está disponível em três apresentações:
- A apresentação de 500 U de FEIBA contém 500 U (unidades) de complexo coagulante antiinibidor em 200 – 600 mg de proteína plasmática humana.
- A apresentação de 1 000 U de FEIBA contém 1 000 U (unidades) de complexo coagulante antiinibidor em 400 – 1 200 mg de proteína plasmática humana.
- A apresentação de 2 500 U de FEIBA contém 2 500 U (unidades) de complexo coagulante antiinibidor em 1 000 – 3 000 mg de proteína plasmática humana.
FEIBA também contém os fatores II, IX e X, principalmente não ativados, assim como fator VII ativado. O antígeno do fator VIII coagulante (FVIII C:Ag) assim como os fatores do sistema de calicreína-cinina estão presentes apenas em quantidades traços.
- Os demais componentes são cloreto de sódio e citrato de sódio.
Diluente
- Água para preparações injetáveis.
Aspecto do produto e conteúdo do envase
O produto se apresenta como um pó liofilizado ou sólido friável, de cor branca a esbranquiçada ou verde pálida. A solução reconstituída tem um pH entre 6,5 e 7,3.
O pó e o diluente são fornecidos em frascos de vidro fechados com tampas de borracha.
Apresentação:1 x 500 U
1 x 1 000 U
1 x 2 500 U
Pode ser que apenas alguns tamanhos de envases sejam comercializados.
Conteúdo do envase:
- 1 frasco com 500 U/1 000 U/2 500 U de FEIBA - pó para solução para perfusão
- 1 frasco com 5 ml/10 ml/25 ml de água para preparações injetáveis
- 1 equipamento para reconstituição BAXJECT II Hi-Flow
- 1 seringa descartável
- 1 agulha borboleta
Título da autorização de comercialização e responsável pela fabricação
Título da autorização de comercialização:
Baxalta Innovations GmbH
Industriestrasse, 67
1221 Viena, Áustria
Responsável pela fabricação:
Takeda Manufacturing Austria AG
Industriestrasse, 67
1221 Viena, Áustria
Representante local do título
Takeda Farmacêutica España, S.A.
Rua Albacete, 5, andar 9º,
Edifício Los Cubos
28027 Madrid
Espanha
Tel: +34 91 790 42 22
Este medicamento está autorizado nos Estados-membros do Espaço Económico Europeu com os seguintes nomes:
Áustria:
FEIBA 100 E./ml Pulver und Lösungsmittel zur Herstellung einer Infusionslösung
Bulgária:
FEIBA 100 U/ml powder and solvent for solution for infusion
Croácia:
FEIBA 100 U/ml prašak i otapalo za otopinu za infuziju
Chipre:
FEIBA 100 U/ml κ?νις και διαλ?της για δι?λυμα προς ?γχυση
República Checa:
FEIBA
Dinamarca:
Feiba
Estônia:
FEIBA 100 Ü/ML
Finlândia:
Feiba
Alemanha:
FEIBA 500 E konzentriert
FEIBA 1000 E konzentriert
FEIBA 2500 E konzentriert
Grécia:
FEIBA 100 U/ml κ?νις και διαλ?της για δι?λυμα προς ?γχυση
Irlanda:
FEIBA 100 U/ml powder and solvent for solution for infusion
Letônia:
Feiba 100 V/ml pulveris un šķidrinātājs infuziju šķidruma pagatavošanai
Lituânia:
Feiba 100 V/ml milteliai ir tirpiklis infuziniam tirpalui
Malta:
FEIBA 100 U/ml powder and solvent for solution for infusion
Países Baixos:
FEIBA 100 E/ML, poeder en oplosmidel voor oplossing voor injectie
Noruega:
Feiba
Romênia:
FEIBA 100 U/ml pulbere si solvent pentru solutie injectabila
Eslováquia:
FEIBA 100 U/ml prášok a rozpúšťadlo na infúzny roztok
Eslovênia:
FEIBA 100 e./ml prašek in vehikel za raztopino za infundiranje
Espanha:
FEIBA 100 U/ml pó e diluente para solução para perfusão
Suécia:
Feiba 100 enheter/ml pulver och vätska till infusionsvätska, lösning
Data da última revisão deste prospecto:08/2024
A informação detalhada deste medicamento está disponível na página web da Agência Espanhola de Medicamentos e Produtos Sanitários (AEMPS) http://www.aemps.gob.es/
---------------------------------------------------------------------------------------------------------------------
Esta informação está destinada unicamente a profissionais do setor sanitário:
O tratamento deve ser iniciado e monitorizado sob a supervisão de um médico com experiência no tratamento dos distúrbios da coagulação.
Posologia
A dose e a duração da terapia dependem da gravidade da alteração da função hemostática, da localização e da gravidade da hemorragia e do estado clínico do paciente.
A dose e a frequência da administração devem ser estabelecidas sempre em função da eficácia clínica em cada caso.
Como guia geral, são recomendadas doses de 50 – 100 U/kg de FEIBA; não deve ser ultrapassada uma dose única de 100 U/kg nem uma dose máxima diária de 200 U/kg, a menos que a gravidade da hemorragia exija e justifique o uso de doses superiores.
Devido a fatores específicos dos pacientes, a resposta a um agente de bypass pode variar, e em uma situação de hemorragia determinada, os pacientes com uma resposta insuficiente a um agente podem responder a outro agente. Em caso de resposta insuficiente a um agente de bypass, deve-se considerar o uso de outro agente.
População pediátrica
A experiência em crianças menores de 6 anos é limitada; deve ser adaptado o mesmo regime posológico que nos adultos para o estado clínico da criança.
- Hemorragia espontânea
Hemorragias em articulações, músculos e tecido mole
Para hemorragias leves a moderadas é recomendada uma dose de 50 – 75 U/kg, cada 12 horas. O tratamento deve continuar até que apareçam claros sinais de melhoria clínica, tais como diminuição da dor, redução do edema ou aumento da mobilização da articulação.
Para hemorragias intensas em músculos e tecido mole, p. ex., hemorragia retroperitoneal, é recomendada uma dose de 100 U/kg cada 12 horas.
Hemorragias em membranas mucosas
É recomendada uma dose de 50 U/kg cada 6 horas, sob estrita vigilância do paciente (controle visual do sangramento, repetição do hematócrito). Se não se detiver a hemorragia, pode-se aumentar a dose para 100 U/kg; no entanto, não deve ser ultrapassada uma dose de 200 U/kg.
Outras hemorragias intensas
Em hemorragia intensa, como hemorragia do SNC, é recomendada uma dose de 100 U/kg cada 12 horas. Em casos particulares, pode-se administrar FEIBA cada 6 horas, até que apareçam claros sinais de melhoria clínica (não deve ser ultrapassada a dose máxima diária de 200 U/kg).
- Cirurgia
Em intervenções cirúrgicas, pode ser administrada uma dose inicial de 100 U/kg antes da operação, e entre as 6 a 12 horas posteriores pode ser administrada outra dose de 50 – 100 U/kg. Como dose de manutenção pós-operatória podem ser administrados 50 – 100 U/kg cada 6 – 12 horas; a dose, os intervalos de dosificação e a duração do tratamento peri- e pós-operatório dependem da intervenção cirúrgica, do estado geral do paciente e da eficácia clínica em cada caso particular (não deve ser ultrapassada a dose máxima diária de 200 U/kg).
- Profilação em pacientes com hemofilia A com inibidor
- Profilação de hemorragias em pacientes com um título alto de inibidor e hemorragias frequentes, após uma resposta falhada frente à indução da imunotolerância (ITI) ou quando dicha indução não se considera:
É recomendada uma dose de 70 – 100 U/kg em dias alternos. Se for necessário, a dose pode ser aumentada para 100 U/kg ao dia ou pode ser reduzida gradualmente.
- Profilação de hemorragias em pacientes com um título alto de inibidor durante a indução da imunotolerância (ITI):
Pode-se administrar FEIBA em combinação com fator VIII, em um intervalo de dose de 50 – 100 U/kg, duas vezes ao dia, até que o título do inibidor do fator VIII tenha diminuído para <2 u.b.*< p>
*1 Unidade Bethesda se define como a quantidade de anticorpos que inibe 50% da atividade do fator VIII em plasma incubado (2 horas a 37°C).
- Uso de FEIBA em grupos especiais de pacientes
FEIBA também foi utilizado em combinação com um concentrado de fator VIII como tratamento de longa duração para a eliminação completa e permanente do inibidor do fator VIII.
Monitorização
Em caso de uma resposta inadequada ao tratamento com o produto, é recomendada a realização de um recuento plaquetário, dado que se considera necessário um número suficiente de plaquetas funcionalmente intactas para que o tratamento com o produto seja eficaz.
Devido ao complexo mecanismo de ação, não se dispõe de uma monitorização direta dos princípios ativos. As provas de coagulação como o tempo de coagulação em sangue total (TCT), o tromboelastograma (TEG, valor r) e o tempo de tromboplastina parcial ativada (TTPa), geralmente mostram apenas pequenos acortamentos, que não se correlacionam necessariamente com a melhoria clínica. Por estas razões, a utilidade destes ensaios para monitorizar o tratamento com FEIBA é muito limitada.
Forma de administração
FEIBA deve ser administrado lentamente, por via intravenosa. FEIBA deve ser perfundido a uma velocidade de perfusão de 2 U/kg/min. Em pacientes que tenham tolerado bem a velocidade de perfusão de 2 U/kg/min, a velocidade de perfusão pode ser aumentada até um máximo de 10 U/kg/min.
FEIBA deve ser reconstituído imediatamente antes de sua administração. A solução deve ser utilizada de imediato (já que a preparação não contém conservantes). Não utilize soluções de aspecto turvo ou que contenham depósitos. A eliminação do medicamento não utilizado e de todos os materiais que tenham estado em contato com ele, será realizada de acordo com a normativa local.
Monitorização da terapia
Não devem ser ultrapassadas doses individuais de 100 U/kg nem doses diárias de 200 U/kg. Os pacientes que recebem mais de 100 U/kg devem ser monitorizados para o desenvolvimento de CID e/ou isquemia coronária aguda e para sintomas de eventos trombóticos ou tromboembólicos. Para deter uma hemorragia devem ser administradas doses altas de FEIBA apenas durante o tempo que seja estritamente necessário.
Se ocorrerem mudanças clinicamente significativas na tensão arterial ou na frequência do pulso, dificuldade respiratória, tosse ou dor no peito, deve-se interromper a perfusão imediatamente e devem ser adotadas as medidas de diagnóstico e terapêuticas necessárias. Os parâmetros analíticos característicos de CID são descenso do fibrinogênio, descenso do recuento de trombócitos e/ou presença de produtos de degradação da fibrina ou do fibrinogênio (PDF). Outros parâmetros para o desenvolvimento de CID são uma clara prolongação do tempo de trombina, do tempo de protrombina ou do tempo de tromboplastina parcial ativada TTPa. Em pacientes com hemofilia com inibidor ou com inibidores adquiridos dos fatores VIII, IX e/ou XI, o TTPa se encontra prolongado devido à doença subjacente.
A administração de FEIBA em pacientes com inibidor pode produzir um incremento inicial “anamnésico” dos níveis do inibidor. Durante a administração contínua de FEIBA, os níveis de inibidores podem descender ao longo do tempo. Tanto os dados clínicos como os dados publicados sugerem que a eficácia de FEIBA não se reduz.
Os pacientes com hemofilia com inibidor ou com inibidores adquiridos de fatores de coagulação que recebem tratamento com FEIBA podem ser mais propensos a sofrer hemorragia ao mesmo tempo que pode aumentar o risco de trombose.
Provas de laboratório e eficácia clínica
Os ensaios in vitro para controlar a eficácia, tais como TTPa, tempo de coagulação total (TCT) e tromboelastograma (TEG) não têm necessariamente uma correlação com a melhoria clínica. Por esta razão, não se deve buscar a normalização destes valores mediante um incremento de doses de FEIBA e até são fortemente rejeitados devido ao possível risco de aparecimento de CID por sobredose.
Importância do recuento plaquetário
Em caso de resposta inadequada ao tratamento com FEIBA, é recomendada a realização de um recuento plaquetário, dado que se considera necessário um número suficiente de plaquetas funcionalmente intactas para que o tratamento com FEIBA seja eficaz.
Tratamento de pacientes com hemofilia B com inibidor
A experiência em pacientes com hemofilia B com inibidor do fator IX é limitada devido à rareza da doença. Cinco pacientes com hemofilia B com inibidor foram tratados com FEIBA durante os ensaios clínicos realizados, ou com tratamento a demanda ou com tratamento profilático ou por intervenções cirúrgicas:
Em um estudo clínico prospectivo, aberto, aleatorizado e paralelo em pacientes com hemofilia A ou B com título de inibidor constantemente elevado (090701, PROOF), se aleatorizou 36 pacientes para receber tratamento profilático ou a demanda, durante 12 meses ± 14 dias. Os 17 pacientes do grupo com tratamento profilático receberam 85 ± 15 U/kg de FEIBA, administrado a dias alternos e os 19 pacientes do grupo com tratamento a demanda receberam tratamento individual, determinado pelo médico. Dois pacientes com hemofilia B com inibidor receberam tratamento a demanda e um paciente com hemofilia B recebeu tratamento profilático. A mediana da Taxa Anual de Hemorragias (TAH) para todos os tipos de episódios hemorrágicos nos pacientes do grupo com tratamento profilático (mediana da TAH = 7,9) foi inferior à dos pacientes do grupo com tratamento a demanda (mediana da TAH = 28,7) o que supõe uma diminuição de 72,5% da mediana da TAH entre os grupos de tratamento.
Em outro estudo prospectivo finalizado, de vigilância de segurança ou não intervencionista do uso perioperatório de FEIBA (PASS-INT-003, SURF), se realizaram um total de 34 intervenções cirúrgicas em 23 pacientes. A maioria dos pacientes (18) padecia hemofilia A congênita com inibidor, dois eram pacientes com hemofilia B com inibidor e três eram pacientes com hemofilia A adquirida com inibidor. O tempo de exposição a FEIBA variou entre 1 e 28 dias, com uma média de 9 dias e uma mediana de 8 dias. A dose média acumulada foi de 88 347 U e a mediana da dose foi de 59 000 U. Nos pacientes com hemofilia B com inibidor, a exposição mais prolongada a FEIBA foi de 21 dias, e a dose máxima administrada foi de 7 324 U.
Também se descreveram na bibliografia 48 pacientes nos quais FEIBA se utilizou para o tratamento e a prevenção de episódios hemorrágicos em pacientes com hemofilia B com inibidor do fator IX (34 pacientes com hemofilia B com inibidor receberam tratamento a demanda, seis pacientes com hemofilia B com inibidor receberam tratamento profilático e oito pacientes com hemofilia B com inibidor receberam tratamento por intervenções cirúrgicas).
Em um estudo cruzado prospectivo, aberto e aleatorizado (091501) se estudaram a tolerabilidade e a segurança de FEIBA reconstituído com um volume regular ou reduzido em 50%, assim como com velocidades de perfusão mais rápidas, em pacientes com hemofilia com inibidor. Se tratou 33 pacientes e 28 pacientes completaram o estudo. Durante o estudo, se reconstituiu FEIBA com um volume reduzido em 50% (concentração de 100 U/ml) e se perfundiu por via intravenosa a velocidades de 2, 4 e 10 U/kg/min à dose indicada de 85 U/kg ± 15 U/kg para todos os pacientes. Os critérios de avaliação primários foram a tolerabilidade e a segurança com o volume reduzido a 50% (maior concentração) e com as velocidades de perfusão estándar e aumentadas. No estudo se demonstrou que tanto a maior concentração (100 U/ml) como as velocidades de perfusão aumentadas (4 e 10 U/kg/min) foram bem toleradas e que o perfil de segurança era comparável ao administrar a dose indicada de 85 U/kg ± 15 U/kg. Os pacientes que receberam o volume reduzido a 50% (maior concentração) à velocidade de perfusão estándar de 2 U/kg/min apresentaram taxas semelhantes de eventos adversos derivados do tratamento (EADT) relacionados que os pacientes que receberam a dose com o volume habitual (concentração de 50 U/ml) e com a mesma velocidade de perfusão. Com a velocidade de perfusão de 4 U/kg/min não se notificaram EADT relacionados. Os pacientes que receberam o volume reduzido a 50% (100 U/ml) à velocidade de perfusão de 10 U/kg/min experimentaram 1 EADT relacionado que não foi grave. Além disso, os pacientes que receberam o volume reduzido a 50% (concentração aumentada) às velocidades de perfusão de 4 e 10 U/kg/min não experimentaram nenhum EADT grave, reações de hipersensibilidade, reações no local de perfusão, EADT trombótico nem EADT que conllevem a interrupção do fármaco nem o abandono do estudo. Em geral, os EADT observados no estudo foram coerentes com o perfil de segurança conhecido de FEIBA em pacientes com hemofilia com inibidor.
Em um estudo de segurança observacional, pós-autorização, aberto, não controlado e não intervencionista de FEIBA (PASS-EU-006), se tratou com FEIBA a 75 pacientes (mediana de idade de 34,8 anos, 70 homens e 5 mulheres), dos quais 73 tinham hemofilia A com inibidor ou hemofilia B com inibidor. Dos 65 pacientes que tinham hemofilia congênita, 63 tinham hemofilia A congênita e 2 tinham hemofilia B congênita. No início, a 43 pacientes se lhes prescreveu FEIBA como tratamento profilático e a 32 pacientes se lhes prescreveu como tratamento a demanda. Se empregaram velocidades de perfusão maiores (> 2 U/kg/min) em 6 pacientes pediátricos com idades compreendidas entre os 11 meses e os 11 anos, e em 5 pacientes adolescentes com idades compreendidas entre os 13 e os 16 anos. Das 320 perfusões realizadas a uma velocidade de perfusão disponível em 7 pacientes pediátricos e em 6 pacientes adolescentes, houve 129 perfusões (40,3%) em 2 pacientes (ambos pediátricos) com uma velocidade de perfusão > 10 U/kg/min, 26 perfusões (8,1%) em 7 pacientes (4 pediátricos; 3 adolescentes) com velocidades de perfusão > 4 e ≤ 10 U/kg/min, 135 perfusões (42,2%) em 7 pacientes (3 pediátricos; 4 adolescentes) com uma velocidade de perfusão > 2 e ≤ 4 U/kg/min, e 30 perfusões (9,4%) em 3 pacientes (1 pediátrico; 2 adolescentes) com uma velocidade de perfusão ≤ 2 U/kg/min.
Além disso, se dispõe de notificações isoladas sobre o uso de FEIBA no tratamento de pacientes com inibidores adquiridos frente aos fatores IX, X, XI e XIII.
Em casos raros, FEIBA também se utilizou em pacientes com presença de inibidor do fator de von Willebrand.
- País de registo
- Substância ativa
- Requer receita médicaSim
- Fabricante
- Esta informação é apenas para referência e não constitui aconselhamento médico. Consulte sempre um médico antes de tomar qualquer medicamento. A Oladoctor não se responsabiliza por decisões médicas baseadas neste conteúdo.
- Alternativas a FEIBA 100 U/mL PÓ E SOLVENTE PARA SOLUÇÃO PARA PERFUSÃOForma farmacêutica: PERFURAÇÃO INJETÁVEL, 50 U/mlSubstância ativa: factor VIII inhibitor bypassing activityFabricante: Baxalta Innovations GmbhRequer receita médicaForma farmacêutica: INJETÁVEL, 1.000 UISubstância ativa: coagulation factor VIIIFabricante: Takeda Manufacturing Austria AgRequer receita médicaForma farmacêutica: INJETÁVEL, 1500 UISubstância ativa: coagulation factor VIIIFabricante: Takeda Manufacturing Austria AgRequer receita médica
Alternativas a FEIBA 100 U/mL PÓ E SOLVENTE PARA SOLUÇÃO PARA PERFUSÃO noutros países
As melhores alternativas com o mesmo princípio ativo e efeito terapêutico.
Alternativa a FEIBA 100 U/mL PÓ E SOLVENTE PARA SOLUÇÃO PARA PERFUSÃO em Польша
Médicos online para FEIBA 100 U/mL PÓ E SOLVENTE PARA SOLUÇÃO PARA PERFUSÃO
Avaliação de posologia, efeitos secundários, interações, contraindicações e renovação da receita de FEIBA 100 U/mL PÓ E SOLVENTE PARA SOLUÇÃO PARA PERFUSÃO – sujeita a avaliação médica e regras locais.









Receba novidades da plataforma e promoções exclusivas
Fique a par das atualizações da Oladoctor e receba promoções exclusivas para subscritores.

