

COFACT 500 IU POLVO Y DISOLVENTE PARA SOLUCION INYECTABLE



Cómo usar COFACT 500 IU POLVO Y DISOLVENTE PARA SOLUCION INYECTABLE
Traducción generada por IA
Este contenido ha sido traducido automáticamente y se ofrece solo con fines informativos. No sustituye la consulta con un profesional sanitario.
Ver originalContenido del prospecto
Introducción
Prospecto: información para el usuario
Cofact 500UI polvo y disolvente para solución inyectable.
Complejo de protrombina humana
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
- Qué es Cofact y para qué se utiliza
- Qué necesita saber antes de empezar a usar Cofact
- Cómo usar Cofact
- Posibles efectos adversos
- Conservación de Cofact
- Contenido del envase e información adicional
1. Qué es Cofact y para qué se utiliza
Cofact contiene como principios activos, los factores II, VII, IX y X, que son factores de coagulación de la sangre humana.
Estos factores son componentes naturales de la sangre humana, y se llaman comúnmente complejo de protrombina. Son dependientes de la vitamina K. Si hay una deficiencia de uno o más de estos factores, pueden ocurrir trastornos de la coagulación sanguínea. Como resultado de esto, hay una mayor tendencia al sangrado. La administración de Cofact ayuda a complementar esta deficiencia, al combatir o prevenir hemorragias.

Cofact puede usarse para:
El tratamiento o prevención de sangrados, debidos a
- deficiencia adquirida de los factores de la coagulación del complejo de protrombina. Por ejemplo, en caso de deficiencia causada por el tratamiento con antagonistas de la vitamina K o en caso de sobredosis de antagonistas de la vitamina K, cuando se requiere una rápida corrección de esta deficiencia.
- deficiencias congénitas de uno de los factores de la coagulación dependientes de la vitamina K, cuando no se dispone de productos purificados del factor de la coagulación específico.
2. Qué necesita saber antes de empezar a usar Cofact

No use Cofact
- si es alérgico (hipersensible) a alguno de los principios activos o a alguno de los demás componentes de este medicamento (incluidos en la sección 6).
Advertencias y precauciones
Consulte a su médico especializado en trastornos de la coagulación antes de recibir este medicamento.
- Si tiene una deficiencia adquirida de los factores de coagulación dependientes de la vitamina K (por ejemplo, causada por el tratamiento con medicamentos antagonistas de la vitamina K), Cofact solo deberá usarse cuando es necesaria una rápida corrección de los niveles del complejo de protrombina, como el caso de un sangrado grave o una intervención quirúrgica de urgencia. En otros casos suele ser suficiente la reducción de la dosis del medicamento antagonista de la vitamina K y/o la administración de vitamina K.
- Si recibe un medicamento antagonista de la vitamina K puede presentar un mayor riesgo de formación de coágulos. En este caso, el tratamiento con Cofact puede potenciar este riesgo.
- Si ha nacido con una deficiencia de alguno de los factores de coagulación dependientes de la vitamina K (deficiencia congénita), deben utilizarse productos específicos para el factor de coagulación cuando estén disponibles.
- Si ocurre una reacción alérgica o de tipo anafiláctico, debe interrumpirse la perfusión con Cofact inmediatamente.
Existe un riesgo de trombosis o coagulación intravascular diseminada (es decir, formación de coágulos de sangre en los vasos sanguíneos) cuando se recibe este medicamento, especialmente cuando se recibe repetidamente.
- Su médico comprobará si la administración de este medicamento representa un riesgo de trombosis (ver sección 4). Las siguientes personas tienen una mayor probabilidad de sufrir una trombosis:
- personas que han sufrido un infarto de miocardio o que han tenido (o todavía tienen) otras enfermedades de las arterias del corazón
- personas con enfermedades del hígado
- recién nacidos (neonatos)
- personas que se van a someter pronto a una operación o que se han operado recientemente
- personas con mayor probabilidad de sufrir complicaciones de coagulación (por ejemplo, antecedentes de acontecimientos tromboembólicos o coagulación intravascular diseminada)
Su médico considerará cuidadosamente el beneficio del tratamiento con este medicamento en comparación con el riesgo de estas complicaciones.
Seguridad vírica
Cuando los medicamentos se preparan a partir de sangre o plasma humanos, se adoptan ciertas medidas para prevenir la transmisión de infecciones a los pacientes. Estas medidas incluyen:
- una cuidadosa selección de los donantes de sangre y plasma para asegurar que se excluye a las personas con riesgo de ser portadores de infecciones,
- el análisis de cada donación y de las mezclas de plasma para detectar la presencia de signos de virus/infecciones,
- la inclusión de etapas en el procesamiento de sangre o plasma que pueden inactivar o eliminar los virus.
A pesar de estas medidas, cuando se administran medicamentos preparados a partir de sangre o plasma humanos, no se puede excluir totalmente la posibilidad de transmisión de infecciones. Esto también se refiere a cualquier virus emergente o de naturaleza desconocida y a otros tipos de infecciones.
Las medidas adoptadas se consideran eficaces para virus envueltos como el virus de la inmunodeficiencia humana (VIH), el virus de la hepatitis B (VHB) y el virus de la hepatitis C (VHC) y para el virus no envuelto de la hepatitis A (VHA). Las medidas adoptadas pueden tener un valor limitado contra otros virus no envueltos como el parvovirus B19. La infección por parvovirus B19 puede ser grave en mujeres embarazadas (infección del feto) y en personas cuyo sistema inmunitario está deprimido o que tienen algún tipo de anemia (p. ej., enfermedad drepanocítica o anemia hemolítica).
Se recomienda encarecidamente que cada vez que se le administre una dosis de este medicamento, se registre el nombre y número de lote del producto, con el fin de tener un historial de los lotes utilizados.
Niños y adolescentes
No se dispone de datos en relación con el uso de este medicamento en niños o adolescentes.
Otros medicamentos y Cofact
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o pudiera tener que utilizar cualquier otro medicamento, incluso los adquiridos sin receta médica.
No se dispone de información relativa a las posibles interacciones entre Cofact y otros medicamentos, a excepción de los anticoagulantes.
Embarazo y lactancia
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de utilizar este medicamento. Si está embarazada o en periodo de lactancia su médico le administrará Cofact solo si está claramente indicado.
Conducción y uso de máquinas
No se han realizado estudios sobre los efectos en la capacidad de conducir o usar máquinas.
Cofact contiene sodio
Cofact contiene hasta 448 mg de sodio (componente principal de la sal de mesa o para cocinar) por 100 ml. Esto equivale a hasta el 22% de la ingesta diaria máxima recomendada de sodio para un adulto. Tenga esto en cuenta si sigue una dieta controlada de sodio.
3. Cómo usar Cofact
Su tratamiento debe ser iniciado, administrado y monitorizado por un médico con experiencia en el tratamiento de los trastornos de la coagulación. Su médico determinará la cantidad de Cofact que usted necesita para el tratamiento o prevención de hemorragias debidas al uso de anticoagulantes o en caso de deficiencia congénita de uno de los factores de la coagulación dependientes de la vitamina K.
La dosis exacta depende de:
- la gravedad de su enfermedad
- su peso
- los factores de la coagulación que usted necesita
- la cantidad de estos factores en su sangre (su nivel en sangre).
En caso de deficiencia congénita de los factores de la coagulación, es importante determinar periódicamente los niveles en sangre de los factores de la coagulación.
La información para profesionales sanitarios se encuentra al final del prospecto.
Si usa más Cofact del que debe
Su médico debe comprobar regularmente el estado de sus coágulos durante el tratamiento. Las dosis elevadas de concentrado de complejo de protrombina se han asociado a casos de infarto de miocardio, coagulación intravascular diseminada y un aumento de la formación de coágulos en un vaso sanguíneo en pacientes con riesgo de sufrir estas complicaciones.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Se han observado los siguientes efectos adversos:
Frecuentes(puede afectar a menos de 1 de cada 10 personas):
- existe un riesgo de formación de coágulos sanguíneos (ver sección 2)
Poco frecuentes(puede afectar a menos de 1 de cada 100 personas):
- existe un riesgo de descenso de la presión arterial
La frecuencia de los siguientes efectos secundarios es desconocida(la frecuencia no puede estimarse a partir de los datos disponibles):
- Reacciones de hipersensibilidad o alérgicas (ver sección 2)
- Ataque al corazón
- Náuseas, vómitos
- Enrojecimiento en la localización de una perfusión, irritación en la localización de una perfusión, hinchazón en la localización de una perfusión, malestar
- Aumento temporal de los resultados de las pruebas hepáticas
- Ictus, mareos
- Embolia pulmonar, dificultad para respirar
- Sudoración excesiva, picor en la piel, urticaria, sarpullido
Los pacientes con deficiencia de uno de los factores de la coagulación II, VII, IX o X pueden desarrollar anticuerpos contra estos factores como resultado de usar Cofact. En ese caso, la actividad del producto no será óptima.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o farmacéutico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano: https://www.notificaRAM.es. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Cofact
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en la caja y en la etiqueta del vial después de CAD. La fecha de caducidad es el último día del mes que se indica.
Conservar en nevera (2 ?C – 8 ?C). No congelar. Conservar el vial en el embalaje exterior para protegerlo de la luz.
Cofact puede conservarse a o por debajo de 25 °C hasta seis meses. La fecha en la que el medicamento ha alcanzado la temperatura ambiente deberá anotarse en el envase. Si no se utiliza durante los seis meses conservado a temperatura ambiente, el producto debe desecharse.
Una vez sacado el producto de la nevera, no debe volver a introducirse en ella.
Se ha demostrado la estabilidad del producto disuelto hasta 3 horas a 15 ºC- 25 ºC. Sin embargo, para prevenir la contaminación, el producto disuelto debe utilizarse inmediatamente.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional
Composición de Cofact
- Los principios activos son los factores de la coagulación II, VII, IX y X y otros principios activos son las proteínas C y proteína S.
- Un vial de Cofact 500 UI contiene 500 UI de factor IX; 280-700 UI de factor II; 140-400 UI de factor VII y 280-700 UI de factor X; 222-780 UI de proteína C; 20-160 UI de proteína S.
Tras disolverla en el agua para preparaciones inyectables que se suministra, la solución para inyectables lista para su uso contiene:
- No menos de 14 UI y no más de 35 UI de factor II por ml;
- No menos de 7 UI y no más de 20 UI de factor VII por ml;
- 25 UI de factor IX por ml;
- No menos de 14 UI y no más de 35 UI de factor X por ml;
- No menos de 11 UI y no más de 39 UI de proteína C por ml;
- No menos de 1 UI y no más de 8 UI de proteína S por ml.
Los demás componentes son citrato de sodio, cloruro de sodio y antitrombina.
Aspecto del producto y contenido del envase
Cofact se suministra como polvo y disolvente para una solución inyectable.
Cofact polvo para inyectable es un polvo de color azulado. El disolvente es un líquido claro, incoloro y sin partículas visibles. La solución preparada y lista para la inyección es una solución de color azulado.
Contenido del envase de 500UI
1 vial de polvo de 500 UI
1 vial de 20 ml de agua para preparaciones inyectables
1 dispositivo de transferencia nextaro v
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización y responsable de la fabricación
Prothya Biosolutions Netherlands B.V.
Plesmanlaan 125
NL-1066 CX Ámsterdam
Países Bajos
Este medicamento está autorizado en los estados miembros del Espacio Económico Europeo con los siguientes nombres:
Austria, Bélgica, Finlandia, Francia, Alemania, Islandia, Italia, Luxemburgo, Países Bajos y España: Cofact.
Suecia: Thyaplex
Fecha de la última revisión de este prospecto: Agosto 2024
--------------------------------------------------------------------------------------------------------------------
Esta información está destinada únicamente a profesionales del sector sanitario:
Composición cualitativa y cuantitativa
Cofact se presenta en forma de polvo y disolvente para solución inyectable que contiene el complejo de protrombina humana. El producto contiene nominalmente las siguientes UI de los factores de la coagulación humanos:
Cofact 500 UI | Después de la reconstitución*(UI/ml) | ||
Principios activos | |||
Factor II de la coagulación | 280 – 700 | 14 – 35 | |
Factor VII de la coagulación | 140 – 400 | 7 – 20 | |
Factor IX de la coagulación | 500 | 25 | |
Factor X de la coagulación | 280 – 700 | 14 – 35 | |
Otros principios activos | |||
Proteína C | 222 – 780 | 11 – 39 | |
Proteína S | 20 – 160 | 1 – 8 | |
*Después de la reconstitución con 20ml de agua para preparaciones inyectables.
El contenido total de proteínas por vial de 500 UI es de 260 – 700 mg. La actividad específica del producto es ≥ 0,6 UI/mg, expresada como actividad del factor IX.
La actividad de todos los factores de coagulación, así como de las proteínas C y S (antígenos) se ha determinado conforme a los estándares actuales de la OMS o de la Farmacopea Europea.
Tras la reconstitución, este medicamento contiene 125 – 195 mmol de sodio/l, hasta 89,6 mg de sodio por vial de 500 UI.
Posología y forma de administración
Posología
A continuación se proporcionan solo unas pautas generales de dosificación. El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de los trastornos de la coagulación. La dosis y duración del tratamiento de sustitución dependen de la gravedad del trastorno, de la localización e intensidad del sangrado y del cuadro clínico del paciente.
La posología y frecuencia de la administración deben calcularse de forma individual para cada paciente. Los intervalos de dosificación se deben adaptar a las diferentes semividas circulantes de los distintos factores de la coagulación en el complejo de protrombina. Los requerimientos posológicos individuales solo pueden calcularse basándose en la determinación periódica de los niveles plasmáticos individuales de los factores de la coagulación en cuestión, o del análisis global de los niveles del complejo de protrombina (tiempo de protrombina, INR) y en la monitorización continua de la situación clínica del paciente.
En el caso de intervenciones de cirugía mayor es imprescindible una monitorización precisa del tratamiento de sustitución, por medio de análisis de coagulación (valoraciones del factor de la coagulación específico y/o análisis globales de los niveles de complejo de protrombina).
Sangrado y profilaxis perioperatoria de sangrados durante el tratamiento con antagonistas de la vitamina K:
La dosis dependerá del INR antes del tratamiento, del INR deseado y del peso corporal. En las siguientes tablas se proporcionan dosis aproximadas que se requieren para la normalización del INR a diferentes niveles iniciales de INR.
Las tablas de dosis solo representan las pautas generales de dosificación, lo que no puede sustituir la evaluación individual de la dosis para cada paciente y el estricto control del INR y otros parámetros de coagulación durante el tratamiento.
Dosis recomendadas de Cofact en ml para alcanzar un INR deseado ≤2,1
INR inicial Peso corporal | 7,5 | 5,9 | 4,8 | 4,2 | 3,6 | 3,3 | 3,0 | 2,8 | 2,6 | 2,5 | 2,3 | 2,2 |
50kg | 40 | 40 | 40 | 30 | 30 | 30 | 20 | 20 | X | X | X | X |
60kg | 50 | 50 | 40 | 40 | 30 | 30 | 30 | 20 | X | X | X | X |
70kg | 60 | 50 | 50 | 50 | 40 | 40 | 30 | 30 | X | X | X | X |
80kg | 60 | 60 | 60 | 50 | 50 | 40 | 40 | 30 | X | X | X | X |
90kg | 60 | 60 | 60 | 60 | 50 | 50 | 40 | 30 | X | X | X | X |
100kg | 60 | 60 | 60 | 60 | 60 | 50 | 40 | 40 | X | X | X | X |
Dosis recomendadas de Cofact en ml para alcanzar un INR deseado ≤1,5
INRinicial Peso corporal | 7,5 | 5,9 | 4,8 | 4,2 | 3,6 | 3,3 | 3,0 | 2,8 | 2,6 | 2,5 | 2,3 | 2,2 |
50kg | 60 | 60 | 60 | 50 | 50 | 50 | 40 | 40 | 30 | 30 | 30 | 30 |
60kg | 80 | 70 | 70 | 60 | 60 | 60 | 50 | 50 | 40 | 40 | 40 | 30 |
70kg | 90 | 80 | 80 | 70 | 70 | 70 | 60 | 60 | 50 | 40 | 40 | 40 |
80kg | 100 | 100 | 90 | 90 | 90 | 80 | 80 | 70 | 60 | 50 | 50 | 40 |
90kg | 100 | 100 | 100 | 90 | 90 | 90 | 80 | 80 | 70 | 60 | 50 | 40 |
100kg | 100 | 100 | 100 | 100 | 100 | 90 | 90 | 80 | 70 | 70 | 60 | 50 |
Las dosis se calculan en función de la concentración de factor IX en Cofact, debido a su semivida relativamente corta y bajo rendimiento tras la perfusión comparado con los otros factores de la coagulación en Cofact. Se presupone que una concentración plasmática media de factor IX ?30% es suficiente para alcanzar un INR de ? 2,1 y ? 60% para alcanzar un INR de ? 1,5. Las cantidades calculadas se redondean en múltiplos de 10 ml y se estableció un límite superior de 60 o 100 ml en total (ver las tablas más arriba). Los valores de INR deseados están recomendados por la Federación de Servicios Holandeses de Trombosis y son del mismo orden que las recomendaciones Inglesas y Alemanas.
La corrección del trastorno de la hemostasia inducido por los antagonistas de la vitamina K persiste durante unas 6-8 horas. Sin embargo, los efectos de la vitamina K, si se administra al mismo tiempo, normalmente se consiguen en un plazo de 4-6 horas. Por lo tanto, normalmente no es necesario repetir el tratamiento con complejo de protrombina humana cuando se ha administrado vitamina K.
Debido a que estas recomendaciones son empíricas y que la recuperación y la duración del efecto pueden variar, es obligatoria la monitorización del INR durante el tratamiento.
Sangrado y profilaxis perioperatoria de la deficiencia congénita de cualquiera de los factores de la coagulación dependientes de la vitamina K, cuando no se dispone del producto de factor de la coagulación específico:
El cálculo de la dosis requerida para el tratamiento se basa en el dato empírico de que aproximadamente 1 UI de factor VII o de factor IX por kg de peso corporal aumenta la actividad plasmática del factor VII o IX, respectivamente, en 0,01 UI/ml; y 1 UI de factor II o X por kg de peso corporal aumenta la actividad plasmática del factor II o X en 0,02 y 0,017 UI/ml, respectivamente.
La dosis de un factor específico administrado se expresa en Unidades Internacionales (UI), que están relacionadas con el estándar actual de la OMS para cada factor. La actividad plasmática de un factor de la coagulación específico se expresa, o bien como un porcentaje (relativo al plasma normal) o en Unidades Internacionales (relativas al estándar internacional para el factor de la coagulación específico).
Una Unidad Internacional (UI) de actividad de un factor de la coagulación es equivalente a la cantidad contenida en un ml de plasma humano normal.
Por ejemplo, el cálculo de la dosis requerida de factor X se basa en el dato empírico de que 1 Unidad Internacional (UI) de factor X por kg de peso corporal aumenta la actividad plasmática del factor X en 0,017 UI/ml. La dosis requerida se determina empleando la siguiente fórmula:
Unidades requeridas = peso corporal (kg) x incremento deseado del factorX (UI/ml) x 60
Donde 60 (ml/kg) es el valor recíproco de la recuperación estimada.
Si se conoce la recuperación individual, este valor debe usarse para el cálculo.
Población pediátrica
No se ha establecido la seguridad y eficacia del uso de Cofact en pacientes pediátricos.
Forma de administración
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6. “Precauciones especiales de eliminación y otras manipulaciones”. Cofact debe administrarse por vía intravenosa.
Se recomienda administrar el producto reconstituido a una velocidad de aproximadamente 2 ml por minuto.
Incompatibilidades
Este medicamento no debe mezclarse con otros medicamentos.
Cofact es compatible con material de polipropileno. El tratamiento puede fracasar debido a la adsorción del factor de la coagulación en la superficie interna de otros equipos de inyección/perfusión.
Periodo de validez
3 años.
Tras la reconstitución, se ha demostrado la estabilidad fisicoquímica en uso durante 3 horas a 15 °C – 25 °C. Desde el punto de vista microbiológico, el producto debe utilizarse inmediatamente después de la reconstitución. Si no se utiliza inmediatamente, los tiempos de conservación en uso y las condiciones antes del uso son responsabilidad del usuario.
Precauciones especiales de eliminación y otras manipulaciones
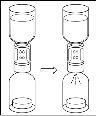
Instrucciones generales para el uso de un dispositivo de transferencia nextarov
- La fracción proteica desecada debe disolverse con 20 ml de agua para preparaciones inyectables. Si los viales cerrados de polvo y disolvente (agua para preparaciones inyectables) se almacenan a 2 ?C – 8 ?C es necesario permitir que alcancen la temperatura ambiente (15 ?C - 25 ?C) antes de disolver la preparación. Esta temperatura se debe mantener durante la reconstitución. Si se utiliza un baño de agua para el calentamiento, se debe tener cuidado para evitar que el agua entre en contacto con los tapones de goma o las tapas abatibles del vial. La temperatura del baño de agua no debe superar los 37 °C.
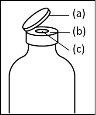
- Durante el procedimiento descrito a continuación se debe aplicar una técnica aséptica. Antes de abrir el envase del dispositivo de transferencia, asegúrese de retirar los tapones abatibles de los viales de polvo y disolvente, y desinfectar el borde del cuello y los tapones de goma con una solución antiséptica y permita que se sequen. No toque los tapones de goma del vial de disolvente ni del vial de polvo.
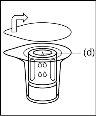
- Como resultado del vacío en el vial de polvo, el disolvente se transfiere automáticamente al vial de polvo.
- Como norma general, el polvo se debe disolver completamente en 10 minutos para formar una solución de color azul. La solución debe ser transparente o ligeramente opalescente. No utilice soluciones turbias o con depósitos. La solución se debe inspeccionar visualmente para detectar partículas y decoloración antes de su administración.
- Cualquier producto no utilizado o material de desecho se debe eliminar de acuerdo con la normativa local.
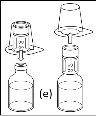
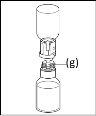
- Procedimiento para el uso de un dispositivo de transferencia nextarov
|
|
|
|
|
|
| |
Nota: el dispositivo de transferencia se debe fijar primero al vial de disolvente y después al vial de polvo. De lo contrario, se producirá una pérdida de vacío y la transferencia del disolvente no se podrá producir. |
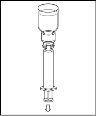
|
Espere hasta que el disolvente se haya transferido completamente. Continúe sujetando la unidad completa formada por el vial de disolvente, el dispositivo de transferencia y el vial de polvo, y asegúrese de que permanece sobre una superficie plana durante todo el proceso de transferencia. Una vez transferido el disolvente, con ambos viales todavía unidos, agite suavemente el vial de polvo hasta que el producto esté completamente disuelto. Para evitar la formación de espuma, no agite el vial. |
|
|
|
|
|
|
|


- País de registro
- Principio activo
- Requiere recetaSí
- Fabricante
- Esta información es de carácter general y no sustituye la consulta con un profesional sanitario.
- Alternativas a COFACT 500 IU POLVO Y DISOLVENTE PARA SOLUCION INYECTABLEForma farmacéutica: INYECTABLE, 1000 ui/vialPrincipio activo: Coagulation factor IX, II, VII and X in combinationFabricante: Csl Behring GmbhRequiere recetaForma farmacéutica: INYECTABLE, 500 UIPrincipio activo: Coagulation factor IX, II, VII and X in combinationFabricante: Csl Behring GmbhRequiere recetaForma farmacéutica: INYECTABLE, 250 UIPrincipio activo: Coagulation factor IX, II, VII and X in combinationFabricante: Prothya Biosolutions Netherlands B.V.Requiere receta
Médicos online para COFACT 500 IU POLVO Y DISOLVENTE PARA SOLUCION INYECTABLE
Comenta la dosis, los posibles efectos secundarios, interacciones, contraindicaciones o la revisión de receta de COFACT 500 IU POLVO Y DISOLVENTE PARA SOLUCION INYECTABLE, sujeto a valoración médica y a la normativa local.









Preguntas frecuentes
