


Smofkabiven extra Nitrogen Ef
Pergunte a um médico sobre a prescrição de Smofkabiven extra Nitrogen Ef



Como usar Smofkabiven extra Nitrogen Ef
Folheto informativo para o utilizador
SmofKabiven extra Nitrogénio EF, emulsão para infusão
Deve ler atentamente o conteúdo do folheto antes de usar o medicamento, pois contém
informações importantes para o doente.
- Deve conservar este folheto para o poder reler se necessário.
- Em caso de dúvidas, deve consultar um médico, farmacêutico ou enfermeiro.
- Se o doente apresentar algum efeito secundário, incluindo quaisquer efeitos secundários não listados neste folheto, deve informar o médico, farmacêutico ou enfermeiro. Ver secção 4.
Índice do folheto
- 1. O que é SmofKabiven extra Nitrogénio EF e para que é utilizado
- 2. Informações importantes antes de usar SmofKabiven extra Nitrogénio EF
- 3. Como usar SmofKabiven extra Nitrogénio EF
- 4. Efeitos secundários possíveis
- 5. Como conservar SmofKabiven extra Nitrogénio EF
- 6. Conteúdo do pacote e outras informações
1. O que é SmofKabiven extra Nitrogénio EF e para que é utilizado
SmofKabiven extra Nitrogénio EF é uma emulsão para infusão, administrada ao doente por gotejamento (infusão intravenosa). O pacote do medicamento é um saco de plástico que contém aminoácidos (componentes necessários para a produção de proteínas), glicose (carboidratos) e gorduras (lípidos). O medicamento pode ser utilizado em doentes adultos e crianças com 2 anos ou mais.
O pessoal médico especializado administra SmofKabiven extra Nitrogénio EF quando outros métodos de nutrição são inadequados ou impossíveis.
2. Informações importantes antes de usar SmofKabiven extra Nitrogénio EF
Não use SmofKabiven extra Nitrogénio EF se o doente tiver:
- alergia aos princípios ativos ou a qualquer um dos outros componentes do medicamento (listados na secção 6);
- alergia a proteínas de peixe ou ovos;
- alergia a amendoins ou soja (SmofKabiven extra Nitrogénio EF contém óleo de soja);
- níveis de gordura no sangue muito elevados (hiperlipidemia);
- disfunção hepática grave;
- problemas de coagulação do sangue (distúrbios da coagulação);
- distúrbio do metabolismo de aminoácidos;
- doença renal grave, sem possibilidade de diálise;
- choque agudo;
- níveis de glicose no sangue não controlados (hiperglicemia);
- líquido nos pulmões (edema pulmonar agudo);
- excesso de líquido no organismo (sobrecarga hídrica);
- insuficiência cardíaca não tratada;
- distúrbio da coagulação do sangue (síndrome hemofagocítica);
- estado geral instável, por exemplo, trauma grave, diabetes não controlada, ataque cardíaco, acidente vascular cerebral, trombose, acidose metabólica (distúrbio caracterizado por uma quantidade excessiva de substâncias ácidas no sangue), infecção grave (sepse grave), coma, desidratação (desidratação hipotônica).
Não use SmofKabiven extra Nitrogénio EF em recém-nascidos e crianças com menos de 2 anos.
Precauções e advertências
Antes de iniciar o tratamento com SmofKabiven extra Nitrogénio EF, deve discutir com o médico se o doente tiver:
- doença renal;
- diabetes;
- pancreatite;
- doença hepática;
- hipotireoidismo (distúrbio da tiróide);
- sepse (infecção grave).
Se durante a infusão ocorrer febre, erupção cutânea, inchaço, dificuldade respiratória, arrepios, suor ou náuseas ou vómitos, deve informar imediatamente o pessoal médico especializado, pois esses sintomas podem ser causados por uma reação alérgica ou dose excessiva do medicamento.
O médico pode recomendar exames de sangue regulares para monitorar a função hepática e outros parâmetros.
Crianças e adolescentes
SmofKabiven extra Nitrogénio EF não é destinado a recém-nascidos ou crianças com menos de 2 anos. SmofKabiven extra Nitrogénio EF pode ser administrado a crianças com 2 a 16/18 anos.
SmofKabiven extra Nitrogénio EF e outros medicamentos
Deve informar o médico sobre todos os medicamentos que o doente está a tomar atualmente ou recentemente, bem como sobre medicamentos que o doente planeia tomar, incluindo aqueles que são vendidos sem receita médica.
Gravidez e amamentação
Não há dados sobre o uso de SmofKabiven extra Nitrogénio EF durante a gravidez ou amamentação. SmofKabiven extra Nitrogénio EF é administrado a mulheres grávidas ou em período de amamentação apenas se o médico considerar necessário. SmofKabiven extra Nitrogénio EF durante a gravidez e amamentação pode ser administrado sob prescrição médica.
Condução de veículos e uso de máquinas
Não se aplica, pois o medicamento é usado em ambiente hospitalar.
3. Como usar SmofKabiven extra Nitrogénio EF
Este medicamento deve ser sempre usado de acordo com as recomendações do médico. Em caso de dúvidas, deve consultar o médico.
O médico escolhe a dose individual com base no peso e estado clínico do doente. SmofKabiven extra Nitrogénio EF é administrado apenas por pessoal médico especializado.
Uso de dose maior do que a recomendada de SmofKabiven extra Nitrogénio EF
É pouco provável que o doente receba uma dose excessiva de SmofKabiven extra Nitrogénio EF, pois o medicamento é administrado por pessoal médico especializado.
4. Efeitos secundários possíveis
Como qualquer medicamento, este medicamento pode causar efeitos secundários, embora não todos os doentes os experimentem.
Efeitos secundários frequentes(podem ocorrer em até 1 em cada 10 doentes):
leve aumento da temperatura corporal.
Efeitos secundários menos frequentes(podem ocorrer em até 1 em cada 100 doentes): níveis elevados de enzimas hepáticas no sangue, perda de apetite, náuseas, vómitos, arrepios, tontura e dor de cabeça.
Efeitos secundários raros(podem ocorrer em até 1 em cada 1000 doentes): pressão arterial baixa ou alta, dificuldade respiratória, ritmo cardíaco acelerado (taquicardia).
Reações de hipersensibilidade (que podem causar sintomas como inchaço, febre, queda da pressão arterial, erupção cutânea, bolhas, vermelhidão, dor de cabeça). Sensação de calor e frio. Palidez. Leve cianose dos lábios e pele (relacionada à hipóxia). Dor no pescoço, nas costas, nos ossos, no peito e na região lombar.
Notificação de efeitos secundários
Se ocorrerem algum efeito secundário, incluindo quaisquer efeitos secundários não listados neste folheto, deve informar o médico, farmacêutico ou enfermeiro. Efeitos secundários podem ser notificados diretamente ao Departamento de Monitorização de Efeitos Secundários de Medicamentos da Agência Reguladora de Medicamentos
Al. Jerozolimskie 181C, 02-222 Warszawa
telefone: +48 22 49 21 301, fax: +48 22 49 21 309
site: https://smz.ezdrowie.gov.pl
Efeitos secundários também podem ser notificados ao titular da autorização de comercialização.
A notificação de efeitos secundários permite reunir mais informações sobre a segurança do medicamento.
5. Como conservar SmofKabiven extra Nitrogénio EF
O medicamento deve ser conservado em local seguro e inacessível a crianças.
Não conservar a uma temperatura superior a 25°C. Não congelar. Conservar no saco exterior.
Não use este medicamento após a data de validade impressa no saco e na caixa de cartão.
A data de validade é o último dia do mês indicado.
6. Conteúdo do pacote e outras informações
O que contém SmofKabiven extra Nitrogénio EF
Os princípios ativos do medicamento são:
g por 1000 ml
alanina
9,2
arginina
7,9
glicina
7,2
histidina
2,0
isoleucina
3,3
leucina
4,8
lisina (como acetato)
4,3
metionina
2,8
fenilalanina
3,3
prolina
7,3
serina
4,3
taurina
0,65
treonina
2,9
triptofano
1,3
tirosina
0,26
valina
4,1
glicose (na forma de composto
monohidratado)
85
óleo de soja refinado
8,7
triglicerídeos de ácidos graxos
saturados de cadeia média
8,7
óleo de oliva refinado
7,2
óleo de peixe rico em ácidos
omega-3
4,3
Os outros componentes (substâncias auxiliares) são: glicerol, fosfolipídios purificados de ovo de galinha, alfa-tocoferol, hidróxido de sódio (para ajustar o pH), oleato de sódio, ácido acético glacial (para ajustar o pH), ácido clorídrico (para ajustar o pH) e água para injeção.
Como é SmofKabiven extra Nitrogénio EF e o que o pacote contém
As soluções de glicose e aminoácidos são transparentes, incolores a ligeiramente amareladas, sem partículas sólidas. A emulsão de gordura é branca e homogênea.
Tamanhos do pacote:
1 × 506 ml, 6 × 506 ml
1 × 1012 ml, 4 × 1012 ml
1 × 1518 ml, 4 × 1518 ml
1 × 2025 ml, 4 × 2025 ml
1 × 2531 ml, 3 × 2531 ml
Nem todos os tamanhos do pacote podem estar disponíveis.
Titular da autorização de comercialização e fabricante
Fresenius Kabi AB
Rapsgatan 7
751 74 Uppsala
Suécia
Para obter informações mais detalhadas, deve contactar o representante do titular da autorização de comercialização:
Fresenius Kabi Polska Sp. z o.o.
Al. Jerozolimskie 134
02-305 Warszawa
telefone: +48 22 345 67 89
Data da última atualização do folheto:09.06.2023
---------------------------------------------------------------------------------------------------------------------------
Informações destinadas apenas ao pessoal médico especializado:
Precauções e advertências para a administração
Para evitar riscos associados à infusão a uma taxa maior do que a recomendada, é recomendável que a infusão seja realizada de forma contínua e controlada, sempre que possível com o uso de uma bomba de volume.
Como o uso de uma veia central para a infusão está associado a um risco aumentado de infecção, durante a colocação e manipulação do cateter, é recomendável seguir rigorosamente as normas de procedimento asséptico para evitar qualquer infecção.
É recomendável monitorar os níveis de glicose e eletrólitos no soro, osmolalidade e balanço de líquidos e equilíbrio ácido-básico, bem como realizar testes enzimáticos hepáticos.
Em caso de ocorrência de qualquer sinal ou sintoma de reação anafilática (como febre, arrepios, erupção cutânea ou dificuldade respiratória), a infusão deve ser interrompida imediatamente.
Não deve ser administrado SmofKabiven extra Nitrogénio EF juntamente com sangue no mesmo conjunto de infusão, devido ao risco de ocorrência de pseudoaglutinação.
Método de administração
Administração intravenosa, infusão em veia central.
Para garantir a nutrição parenteral completa, deve adicionar ao medicamento SmofKabiven extra Nitrogénio EF oligoelementos, eletrólitos e vitaminas, de acordo com as necessidades do doente.
Posologia
Doentes adultos
Dose recomendada
O intervalo de dose é de 13 a 31 ml de SmofKabiven extra Nitrogénio EF/kg de peso corporal/dia, o que fornece de 0,14 a 0,32 g de nitrogênio/kg de peso corporal/dia (de 0,85 a 2,0 g de aminoácidos/kg de peso corporal/dia) e de 12 a 28 kcal/kg de peso corporal/dia de energia total (de 8 a 19 kcal/kg de peso corporal/dia de energia não proteica).
Taxa de infusão
A taxa máxima de infusão de glicose é geralmente de 0,25 g/kg de peso corporal/hora, de aminoácidos 0,1 g/kg de peso corporal/hora e de gorduras 0,15 g/kg de peso corporal/hora.
A taxa de infusão não deve ser superior a 1,5 ml/kg de peso corporal/hora (o que corresponde a 0,13 g de glicose, 0,10 g de aminoácidos e 0,04 g de gorduras/kg de peso corporal/hora). O tempo recomendado de infusão é de 14 a 24 horas.
Nutrição parenteral durante a diálise (IDPN, do inglês intradialytic parenteral nutrition)
Em doentes adultos clinicamente estáveis submetidos a terapia de substituição renal crônica, a taxa máxima de infusão durante a nutrição parenteral durante a diálise (IDPN) é de 3,0 ml/kg de peso corporal/hora (o que corresponde a 0,20 g/kg de peso corporal/hora de aminoácidos, 0,25 g/kg de peso corporal/hora de glicose e 0,09 g/kg de peso corporal/hora de gorduras). O volume de infusão durante a IDPN deve ser baseado na diferença entre a ingestão oral de nutrientes e a ingestão recomendada de nutrientes, perdas inevitáveis de nutrientes devido à terapia de substituição renal, bem como na tolerância metabólica individual do doente. O tempo de infusão durante a IDPN é geralmente de 3 a 5 horas, dependendo das necessidades do doente e do tempo planejado para a sessão de terapia de substituição renal. A dose diária máxima recomendada permanece inalterada.
Dose diária máxima
A dose diária máxima depende do estado clínico do doente e pode variar mesmo de um dia para o outro. A dose diária máxima recomendada é de 31 ml/kg de peso corporal/dia.
Crianças e adolescentes
Crianças de 2 a 11 anos
Dose recomendada
A dose de até 31 ml/kg de peso corporal/dia deve ser ajustada regularmente às necessidades do doente em idade pediátrica, que variam muito mais do que nos doentes adultos.
Taxa de infusão
A taxa máxima de infusão recomendada é de 1,8 ml/kg de peso corporal/hora (o que corresponde a 0,12 g de aminoácidos/kg de peso corporal/hora, 0,15 g de glicose/kg de peso corporal/hora e 0,05 g de gorduras/kg de peso corporal/hora).
Fora de situações especiais que exigem monitoramento cuidadoso, quando a taxa máxima de infusão recomendada for usada, o tempo de infusão não deve exceder 17 horas.
O tempo de infusão recomendado é de 12 a 24 horas.
Dose diária máxima
A dose diária máxima é variável, dependendo do estado clínico do doente, e pode mudar mesmo de um dia para o outro. A dose diária máxima recomendada é de 31 ml/kg de peso corporal/dia.
Adolescentes de 12 a 16/18 anos
Em adolescentes, SmofKabiven extra Nitrogénio EF pode ser administrado como em doentes adultos.
Precauções especiais para a eliminação e preparação do medicamento para uso
Não use se o pacote estiver danificado.
Use apenas se as soluções de aminoácidos e glicose forem transparentes, incolores a ligeiramente amareladas, e a emulsão de gordura for branca e homogênea .O conteúdo das três câmaras separadas deve ser misturado antes do uso, e também antes da adição de outros componentes através do porto designado para esse fim.
Após a remoção das proteções, deve agitar o saco várias vezes para misturar completamente todos os componentes do medicamento e obter uma mistura homogênea, na qual não devem ser visíveis sinais de separação de fases.
Apenas para uso único. Qualquer medicamento não utilizado após a infusão deve ser destruído.
Compatibilidade
Dados sobre a compatibilidade estão disponíveis para os medicamentos Dipeptiven, Addamel N/Supliven, Glycophos, Addiphos, Vitalipid N Adult/Infant e Soluvit N em quantidades específicas e em eletrólitos com concentração específica.
Ao adicionar eletrólitos, deve levar em conta as quantidades já presentes no saco para atender às necessidades clínicas do doente. Dados disponíveis confirmam a possibilidade de adicionar os medicamentos mencionados ao saco ativado, de acordo com a tabela abaixo:
Intervalo de compatibilidade: estável por 7 dias, ou seja, 6 dias armazenado a 2-8 °C, e subsequentemente 24 horas a 20-25 °C.
| Unidade | Quantidade máxima total | |||||
| Tamanho do saco SmofKabiven extra Nitrogénio EF | ml | 506 | 1012 | 1518 | 2025 | 2531 |
| Aditivo | Volume | |||||
| Dipeptiven | ml |
|
|
|
|
|
| Supliven/Addamel N | ml |
|
|
|
|
|
| Soluvit N | ampola(s) |
|
|
|
|
|
| Vitalipid N Adult/Infant | ml |
|
|
|
|
|
| Limites de eletrólitos1 | Concentração | |||||
| Sódio | mmol/l | ≤ 150 | ≤ 150 | ≤ 150 | ≤ 150 | ≤ 150 |
| Potássio | mmol/l | ≤ 150 | ≤ 150 | ≤ 150 | ≤ 150 | ≤ 150 |
| Cálcio | mmol/l | ≤ 5 | ≤ 5 | ≤ 5 | ≤ 5 | ≤ 5 |
| Magésio | mmol/l | ≤ 5 | ≤ 5 | ≤ 5 | ≤ 5 | ≤ 5 |
| Fosfato inorgânico (Addiphos) ou Fosfato orgânico (Glycophos) | mmol/l | ≤ 15 ≤ 30 | ≤ 15 ≤ 30 | ≤ 15 ≤ 30 | ≤ 15 ≤ 30 | ≤ 15 ≤ 30 |
| Zinco | mmol/l | ≤ 0,2 | ≤ 0,2 | ≤ 0,2 | ≤ 0,2 | ≤ 0,2 |
| Selênio | µmol/l | ≤ 2 | ≤ 2 | ≤ 2 | ≤ 2 | ≤ 2 |
Observação: esta tabela tem como objetivo mostrar a compatibilidade. Não constitui diretrizes para a posologia.
Antes de prescrever os medicamentos mencionados, deve consultar os folhetos de informação aprovados.
Informações sobre a compatibilidade com outros aditivos e tempos de armazenamento de diferentes misturas estarão disponíveis a pedido.
Qualquer aditivo deve ser misturado com o medicamento em condições assépticas.
Prazo de validade após a mistura do conteúdo das câmaras do saco
Foi demonstrada a estabilidade física e química do conteúdo misturado do saco tricameral por 48 horas a 20-25 °C. Do ponto de vista microbiológico, o medicamento deve ser usado imediatamente.
Em caso contrário, o prazo de validade durante o uso e as condições de armazenamento antes da administração são de responsabilidade do usuário. Este prazo não deve, em princípio, exceder 24 horas a 2-8 °C, a menos que a mistura tenha sido feita em condições assépticas controladas e validadas.
Prazo de validade após a mistura com substâncias adicionais
Foi demonstrada a estabilidade físico-química do conteúdo misturado do saco tricameral com substâncias adicionais por um período de até 7 dias, ou seja, 6 dias a 2-8 °C, e subsequentemente 24 horas a 20-25 °C, incluindo o tempo de infusão. Do ponto de vista microbiológico, o medicamento deve ser usado imediatamente após a adição de outros componentes. Em caso contrário, o prazo de validade durante o uso e as condições de armazenamento antes da administração são de responsabilidade do usuário. Este prazo não deve, em princípio, exceder 24 horas a 2-8 °C, a menos que a mistura tenha sido feita em condições assépticas controladas e validadas.
SmofKabiven extra Nitrogénio EF Instruções para a preparação do saco para uso
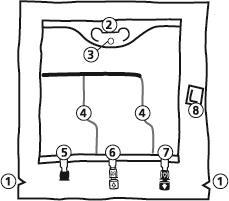
- 1. Corte no saco exterior
- 2. Alça do saco
- 3. Orifício para suspensão do saco
- 4. Soldas que separam as câmaras do saco
- 5. Porto cego (usado apenas na produção)
- 6. Porto para administração de substâncias adicionais
- 7. Porto de infusão
- 8. Absorvedor de oxigênio
| |
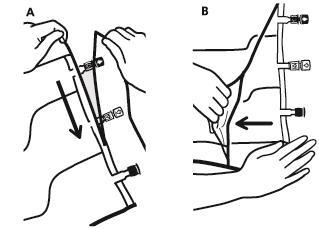 | |
- Para remover o saco exterior, deve colocá-lo na horizontal e, começando pelo corte que se encontra perto dos portos, rasgar ao longo da borda superior (A).
- Em seguida, rasgar o saco exterior ao longo da borda longa, remover e descartar juntamente com o absorvedor de oxigênio (B).
2. Mistura
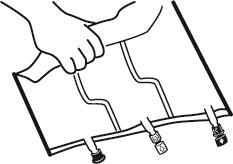
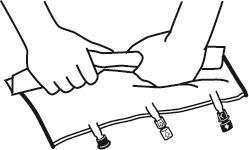
- Colocar o saco em uma superfície plana.
- Começando pela alça, rolar firmemente o saco em direção aos portos, primeiro com a mão direita e, em seguida, aplicando pressão constante com a mão esquerda, até que as soldas verticais se abram. Elas se abrem sob a pressão do líquido. As soldas também podem ser abertas antes da remoção do saco exterior. Observação:o líquido mistura-se facilmente, embora a solda horizontal permaneça intacta.
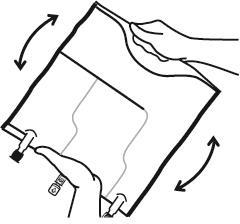
- Misturar o conteúdo das três câmaras virando o saco três vezes, o que deve garantir a mistura completa dos componentes.
| |
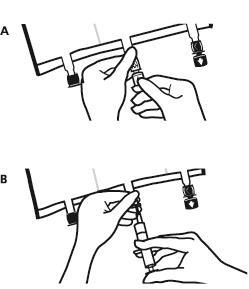 | |
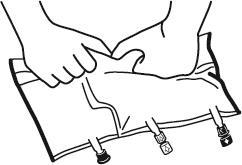
- Colocar o saco novamente em uma superfície plana e nivelada. Imediatamente antes da administração de substâncias adicionais, remover a tampa marcada com uma seta que protege o porto branco para administração de substâncias adicionais (A).
Observação:a membrana do porto para administração de substâncias adicionais é estéril.
- Segurar a base do porto para administração de substâncias adicionais. Introduzir a agulha, injetar as substâncias adicionais (com compatibilidade conhecida) através do centro do local de injeção (B).
- Misturar cuidadosamente o conteúdo do saco após a adição de cada componente, girando o saco três vezes após cada adição. Use seringas com agulhas de diâmetro 18 a 23 G e comprimento máximo de 40 mm.
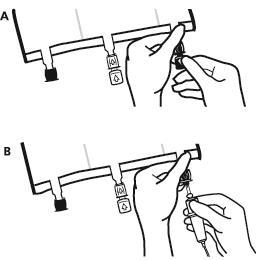
- Imediatamente antes de conectar o conjunto de infusão, remover a tampa de uso único que protege o porto de infusão azul (A). Observação:a membrana do porto de infusão é estéril.
- Use conjuntos de infusão sem detector de ar ou feche o detector de ar.
- Segurar a base do porto de infusão.
- Introduzir a agulha do conjunto de infusão no porto de infusão. Para garantir uma fixação segura da agulha, deve inserir toda a sua extensão. Observação:a superfície interna do porto de infusão é estéril.
4. Suspensão do saco
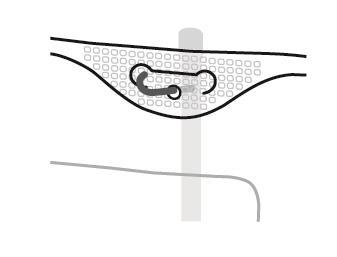
- Suspender o saco usando o orifício localizado abaixo da alça.
- País de registo
- Substância ativa
- Requer receita médicaSim
- Fabricante
- ImportadorFresenius Kabi AB
- Esta informação é apenas para referência e não constitui aconselhamento médico. Consulte sempre um médico antes de tomar qualquer medicamento. A Oladoctor não se responsabiliza por decisões médicas baseadas neste conteúdo.
- Alternativas a Smofkabiven extra Nitrogen EfForma farmacêutica: Solução, -Substância ativa: combinationsNão requer receita médicaForma farmacêutica: Solução, -Substância ativa: combinationsNão requer receita médicaForma farmacêutica: Solução, -Substância ativa: combinationsNão requer receita médica
Alternativas a Smofkabiven extra Nitrogen Ef noutros países
As melhores alternativas com o mesmo princípio ativo e efeito terapêutico.
Alternativa a Smofkabiven extra Nitrogen Ef em Espanha
Alternativa a Smofkabiven extra Nitrogen Ef em Ukraine
Médicos online para Smofkabiven extra Nitrogen Ef
Avaliação de posologia, efeitos secundários, interações, contraindicações e renovação da receita de Smofkabiven extra Nitrogen Ef – sujeita a avaliação médica e regras locais.









Receba novidades da plataforma e promoções exclusivas
Fique a par das atualizações da Oladoctor e receba promoções exclusivas para subscritores.

