

KYBERNIN P 1000 UI, POLVO Y DISOLVENTE PARA SOLUCION PARA INYECCION O PERFUSION



Cómo usar KYBERNIN P 1000 UI, POLVO Y DISOLVENTE PARA SOLUCION PARA INYECCION O PERFUSION
Traducción generada por IA
Este contenido ha sido traducido automáticamente y se ofrece solo con fines informativos. No sustituye la consulta con un profesional sanitario.
Ver originalContenido del prospecto
Introducción
Prospecto: información para el usuario
Kybernin P 1000 UI polvo y disolvente para solución inyectable y para perfusión.
Antitrombina III humana
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento,porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
- Qué es Kybernin P y para qué se utiliza
- Qué necesita saber antes de empezar a usar Kybernin P
- Cómo usar Kybernin P
- Posibles efectos adversos
- Conservación de Kybernin P
- Contenido del envase e información adicional
1. Qué es Kybernin P y para qué se utiliza
Kybernin P es un polvo y disolvente para solución inyectable y para perfusión.
Este medicamento pertenece al grupo de medicamentos llamados agentes antitrombóticos.
Kybernin P se utiliza si usted tiene un déficit congénito de antitrombina, para prevenir la formación y el desarrollo de coágulos en los vasos sanguíneos de sus piernas (trombosis venosa profunda) o en otros vasos de su cuerpo (tromboembolismo) durante la cirugía o en el periodo peri-parto y en asociación con heparina si está indicado.
Kybernin P también se utiliza si usted tiene déficit adquirido de antitrombina.

2. Qué necesita saber antes de empezar a usar Kybernin P
No use Kybernin P:
Si es alérgico al principio activo o a alguno de los demás componentes de este medicamento (incluidos en la sección 6).
Advertencias y precauciones
Al igual que con cualquier producto proteico para administración intravenosa, es posible que se produzcan reacciones de hipersensibilidad de tipo alérgico. Se requiere una estrecha monitorización y cuidadosa observación de los pacientes para detectar cualquier síntoma durante el periodo de perfusión. Se debe informar a los pacientes acerca de los signos iniciales de las reacciones de hipersensibilidad, que incluyen erupciones cutáneas que pueden llegar a urticaria generalizada, opresión torácica, dificultad al respirar, hipotensión y anafilaxia. Si se producen estos síntomas después de la administración, contactar con el médico.
En caso de shock, se seguirán las recomendaciones vigentes para el tratamiento del mismo.
Seguridad viral
Cuando se administran medicamentos derivados de sangre o plasma humanos, hay que llevar a cabo ciertas medidas para evitar que las infecciones pasen a los pacientes. Tales medidas incluyen:
- Una cuidadosa selección de los donantes, para excluir a aquellos que están en riesgo de ser portadores de enfermedades infecciosas,
- Análisis de marcadores específicos de infecciones en las donaciones individuales y en las mezclas de plasma,
- La inclusión de etapas en el proceso de fabricación para eliminar/inactivar virus.
A pesar de esto, cuando se administran medicamentos derivados de la sangre o plasma humanos, la posibilidad de transmisión de agentes infecciosos no se puede excluir totalmente. Esto también se refiere a virus emergentes o de naturaleza desconocida u otros tipos de infecciones.
Las medidas tomadas se consideran efectivas para virus envueltos tales como el virus de la inmunodeficiencia humana (VIH), el virus de la hepatitis B (VHB), el virus de la hepatitis C (VHC), y para los virus no envueltos tales como la hepatitis A (VHA) y el parvovirus B19.
Su médico puede recomendarle que considere la vacunación contra hepatitis A y B si recibe regularmente productos con antitrombina derivada de plasma humano.
Es altamente recomendable que cada vez que se administre Kybernin P a un paciente se deje constancia del nombre del medicamento y nº de lote administrado a fin de mantener una relación entre el paciente y el lote de producto.
Monitorización clínica y biológica en caso de administración conjunta de antitrombina y heparina:
- A fin de ajustar la dosis de heparina y evitar una excesiva hipocoagulabilidad, se deben realizar regularmente controles del alcance de la anticoagulación (APPT, y cuando proceda actividad anti-FXa), a intervalos cortos y en especial en los primeros minutos/horas posteriores al inicio de la administración de la antitrombina.
- Determinación diaria de los niveles de antitrombina, a fin de ajustar la dosis individual, debido al riesgo de disminución de los niveles de antitrombina como consecuencia de un tratamiento prolongado con heparina no fraccionada.
Uso de Kybernin P con otros medicamentos
Heparina: la reposición de antitrombina durante la administración de heparina en dosis terapéuticas aumenta el riesgo de sangrado. El efecto de la antitrombina se ve potenciado en gran medida por la heparina. La semivida de la antitrombina puede disminuir considerablemente por el tratamiento concomitante con heparina, debido a la movilización acelerada de la antitrombina. Por consiguiente, la administración simultánea de heparina y antitrombina a un paciente con riesgo elevado de sangrado se debe monitorizar clínica y biológicamente.
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o pudiera tener que utilizar cualquier otro medicamento.
Embarazo, lactancia y fertilidad
La experiencia en relación con la seguridad de los productos de antitrombina humana para su uso en embarazo humano es limitada.
La seguridad de uso de Kybernin P en el embarazo humano no se ha establecido en ensayos clínicos controlados. Los estudios en animales de experimentación son insuficientes para valorar la seguridad en relación con la reproducción, desarrollo del embrión o del feto, el curso de la gestación y el desarrollo peri y postnatal.
No hay experiencias negativas en relación al tratamiento durante el embarazo y la lactancia.
Por lo tanto, Kybernin P debe administrarse a mujeres embarazadas o lactantes con déficit de antitrombina solamente si está claramente indicado, teniendo en cuenta que el embarazo confiere un aumento del riesgo de episodios tromboembólicos en estas pacientes.
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de utilizar este medicamento. Su médico sopesará el posible riesgo para el feto y le informará si el tratamiento con este medicamento es adecuado. Su médico le recomendará este tratamiento únicamente si está claramente indicado.

Conducción y uso de máquinas
No existe ningún indicio de que Kybernin P pueda afectar la capacidad para conducir vehículos o manejar maquinaria.
KyberninP contiene sodio
Los pacientes con dietas pobres en sodio deben tener en cuenta que Kybernin P 1000 UI contiene hasta 89,52 mg (3,894 mmol) de sodio por 1.000 UI.
3. Cómo usar Kybernin P
Kybernin P es un medicamento de uso hospitalario, por lo que se le administrará en un
hospital por el personal sanitario correspondiente.
Kybernin P se administra preparando una solución previa, la cual se inyecta o perfunde por
vía intravenosa lentamente (máximo 4 ml/min).
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico o farmacéutico. En caso de duda, consulte de nuevo a su médico o farmacéutico.
Su médico indicará con qué frecuencia y a qué intervalos debe administrarse Kybernin P.
Su médico le indicará la duración de su tratamiento con Kybernin P.
Sí usa más Kybernin P del que debe:
No se han notificado síntomas de sobredosificación con antitrombina.
En caso de sobredosis o administración accidental, consultar al Servicio de Información Toxicológica. Teléfono 91 562 04 20.
Si olvidó usar Kybernin P:
- Consulte inmediatamente a su médico o farmacéutico.
- No administrar una dosis doble para compensar las dosis olvidadas.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Las siguientes reacciones adversas se basan en la experiencia poscomercialización. En los casos en los que se dispone de datos se han utilizado las siguientes categorías estándar de frecuencia:
Muy frecuente >1/10
Frecuente >1/100 a <1/10
Poco frecuentes ≥ 1/1.000 a < 1/100
Raras ≥ 1/10.000 a < 1/1.000
Muy raras < 1/10.000 (incluyendo los casos individuales reportados)
Clasificación por Órganos y Sistemas | Término Preferido | Frecuencia |
Trastornos del sistema inmunológico | Hipersensibilidad/reacciones anafilácticas, incluyendo anafilaxis grave y shock. | Rara |
Trastornos generales y en el lugar de administración | Pirexia | Rara |
Para información sobre seguridad viral, ver “Advertencias y precauciones” en la sección 2 de este prospecto.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano: https://www.notificaram.es. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Kybernin P
Mantener este medicamento fuera de la vista y del alcance de los niños.
No conservar a temperatura superior a 25 ºC. No congelar.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase después de EXP. La fecha de caducidad es el último día del mes que se indica.
No usar soluciones que estén turbias o presenten residuos (depósitos/partículas).
Después de la reconstitución, la estabilidad físico-química ha sido demostrada para un tiempo de 8 horas a temperatura ambiente (máx. 25 ºC). Desde un punto de vista microbiológico y dado que Kybernin P no contiene conservantes, la solución reconstituida debe usarse inmediatamente. Si ello no es posible, no almacenar más de 8 horas a temperatura ambiente (máximo 25 ºC).
La eliminación del medicamento no utilizado o del material de desecho, se realizará de acuerdo a las normativas locales.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional
Composición de Kybernin P 1.000 UI
- El principio activo es antitrombina III. Cada vial liofilizado contiene 1.000 UI de antitrombina III. La solución reconstituida contiene aproximadamente 50 UI de antitrombina III/ml de antitrombina derivada de plasma humano cuando se reconstituye con 20 ml de agua para preparaciones inyectables.
La potencia (UI) se determina utilizando el método del Sustrato Cromogénico de acuerdo a la Farmacopea Europea. La actividad específica de Kybernin P es aproximadamente 5,3 UI/mg de proteína.
- Los demás componentes son: glicina, cloruro sódico, citrato sódico, ácido clorhídrico o hidróxido sódico (para ajustar el pH) y agua para preparaciones inyectables.
Ver sección 2 para información importante sobre alguno de los excipientes.
Aspecto del producto y contenido del envase
Polvo y disolvente para solución inyectable y para perfusión.
El envase de comercialización contiene un vial de inyección de vidrio moldeado tipo II (según
Farm. Eur.), incoloro y sellado con un tapón de caucho, disco de plástico y cápsula de aluminio
conteniendo el liofilizado, un vial con 20 ml de agua para preparaciones inyectables (disolvente
para la preparación de la solución) y un trasvasador.
Presentaciones:
Envase individual de Kybernin P 1.000 UI:
1 vial de liofilizado
1 vial con 20 ml de agua para inyectables
1 trasvasador
Envase clínico de Kybernin P 1.000 UI:
10 viales de liofilizado
10 viales con 20 ml de agua para preparaciones inyectables
10 trasvasadores
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización y responsable de la fabricación
Titular de la autorización de comercialización
CSL Behring, S.A.
c/ Tarragona 157, planta 18
08014 Barcelona - España
Responsable de la fabricación
CSL Behring GmbH
Emil-von-Behring-Str. 76
35041 Marburg - Alemania
Fecha de la última revisión de este prospecto: Noviembre 2020
La información detallada y actualizada de este medicamento está disponible en la página web de la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) http://www.aemps.gob.es
Esta información está destinada únicamente a profesionales del sector sanitario:
Dosificación
En el déficit congénito, la dosis debe individualizarse para cada paciente, teniendo en cuenta la historia familiar por lo que respecta a episodios tromboembólicos, los factores de riesgo clínico del paciente y las pruebas de laboratorio.
La dosificación y duración de la terapia de sustitución en el déficit adquirido dependen del nivel de antitrombina plasmática, la presencia de signos de movilización aumentada, el trastorno subyacente y la gravedad del cuadro clínico del paciente. La dosis y la frecuencia de administración deben basarse siempre en la eficacia clínica y en las pruebas de laboratorio en cada caso en particular.
El número de unidades de antitrombina administradas se expresa en Unidades Internacionales (UI), en relación con el estándar de la Organización Mundial de la Salud (OMS) vigente para la antitrombina. La actividad plasmática de antitrombina se expresa como un porcentaje (en relación con el plasma humano normal) o en Unidades Internacionales (en relación con un estándar internacional para antitrombina en plasma).
Una unidad internacional (UI) de actividad de antitrombina es equivalente a la cantidad de antitrombina en 1 ml de plasma humano normal. El cálculo de la dosis necesaria de antitrombina se basa en el hallazgo empírico de que 1 Unidad Internacional (UI) de antitrombina por kg de peso corporal eleva la actividad de antitrombina plasmática en aproximadamente un 1,5%.
La dosis inicial se determina mediante la fórmula siguiente:
Unidades requeridas = peso corporal [kg] x (100 – actividad actual de la antitrombina [%]) x 2/3
La actividad de antitrombina que debe alcanzarse inicialmente depende del estado clínico. Cuando se establece que la sustitución con antitrombina está indicada, la dosis debería ser la suficiente para alcanzar la actividad de antitrombina deseada y para mantener un nivel efectivo. La dosis debe determinarse y monitorizarse de acuerdo a las pruebas de laboratorio de la actividad de antitrombina, que se realizarán, al menos, dos veces al día hasta la estabilización del paciente, posteriormente una vez al día, preferentemente inmediatamente antes de la siguiente perfusión. El ajuste de la dosis debe tener en cuenta tanto los signos de producción aumentada de antitrombina de acuerdo con las pruebas de laboratorio como la evolución clínica. La actividad de antitrombina debe mantenerse por encima del 80% durante el tratamiento, a no ser que el estado clínico indicase un nivel de eficacia diferente.
La dosis inicial habitual en deficiencia congénita es de 30-50 UI/kg.
Por lo tanto, la dosis y la frecuencia de administración, así como la duración del tratamiento deben ajustarse a los datos biológicos y situación clínica.
Población pediátrica:
Kybernin P no está recomendado para uso en niños menores de 6 años debidos a la escasez de datos.
En base a la experiencia clínica, no puede recomendarse el uso de antitrombina en el tratamiento del SDRI (Síndrome de distrés respiratorio infantil) en niños prematuros.
Instrucciones para la correcta administración del preparado
Instrucciones generales
El polvo liofilizado debe reconstituirse completamente, bajo condiciones asépticas, con el disolvente acompañante. Se obtiene una solución transparente o ligeramente opalescente.
El diluyente apropiado es una solución de albúmina humana al 5%. Para preparar diluciones de una titulación de hasta 1:5, pueden utilizarse también: solución Ringer lactato, solución salina fisiológica, solución de glucosa al 5% o poligelina.
El uso de almidones hidroxietilados no se recomiendan como disolvente (para perfusión), ya que se ha observado una pérdida de actividad de antitrombina.
Este medicamento no debe mezclarse con otros medicamentos en la jeringa/equipo de perfusión. Dopamina, dobutamina y furosemida no deben administrarse por el mismo acceso venoso.
El producto debe administrarse por vía intravenosa. Velocidad de perfusión máxima: 4 ml/min.
Reconstitución
Para un manejo correcto del Transofix® de doble punta, siga los pasos a continuación:
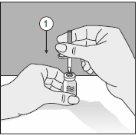
- Después de retirar una de las dos tapas de protección, inserte la punta descubierta perpendicularmente en el tapón de goma del vial de disolvente.
- Retire la tapa de protección de la segunda punta.
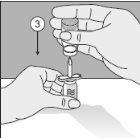
- Perfore la cabeza del vial del producto en esta punta.
- Gire toda la unidad 180°.
- Colóquelo de manera que la base del vial de producto quede apoyada en la superficie de la mesa. El solvente fluye ahora al vial del producto.
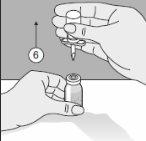
- El Transofix® de doble punta junto con el vial de disolvente se extrae del vial del producto y, posteriormente, se disuelve Kybernin P. Kybernin P reconstituido puede extraerse en una jeringa y administrarse.
- País de registro
- Principio activo
- Requiere recetaSí
- Fabricante
- Esta información es de carácter general y no sustituye la consulta con un profesional sanitario.
- Alternativas a KYBERNIN P 1000 UI, POLVO Y DISOLVENTE PARA SOLUCION PARA INYECCION O PERFUSIONForma farmacéutica: INYECTABLE PERFUSION, 1.000 Ul. antitrombina III humanaPrincipio activo: Antitrombina iiiFabricante: Octapharma S.A.Requiere recetaForma farmacéutica: INYECTABLE PERFUSION, 500 U.l. antitrombina III humanaPrincipio activo: Antitrombina iiiFabricante: Octapharma S.A.Requiere recetaForma farmacéutica: INYECTABLE, 500 UI antitrombinaPrincipio activo: Antitrombina iiiFabricante: Csl Behring S.A.Requiere receta
Médicos online para KYBERNIN P 1000 UI, POLVO Y DISOLVENTE PARA SOLUCION PARA INYECCION O PERFUSION
Comenta la dosis, los posibles efectos secundarios, interacciones, contraindicaciones o la revisión de receta de KYBERNIN P 1000 UI, POLVO Y DISOLVENTE PARA SOLUCION PARA INYECCION O PERFUSION, sujeto a valoración médica y a la normativa local.









Preguntas frecuentes
