

FEIBA 100 U/ML POLVO Y DISOLVENTE PARA SOLUCIÓN PARA PERFUSIÓN




Cómo usar FEIBA 100 U/ML POLVO Y DISOLVENTE PARA SOLUCIÓN PARA PERFUSIÓN
Traducción generada por IA
Este contenido ha sido traducido automáticamente y se ofrece solo con fines informativos. No sustituye la consulta con un profesional sanitario.
Ver originalContenido del prospecto
Introducción
Prospecto: información para el usuario
FEIBA 100U/ml polvo y disolvente para solución para perfusión
complejo coagulante antiinhibidor
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
- Qué es FEIBA y para qué se utiliza
- Qué necesita saber antes de empezar a usar FEIBA
- Cómo usar FEIBA
- Posibles efectos adversos
- Conservación de FEIBA
- Contenido del envase e información adicional
1. Qué es FEIBA y para qué se utiliza
FEIBA es una preparación elaborada a partir de plasma humano, que permite la hemostasia, incluso cuando la cantidad de los factores de coagulación específicos está reducida o ausente.
FEIBA se utiliza para el tratamiento y la profilaxis de hemorragias en pacientes con hemofilia A con inhibidor.
FEIBA se utiliza para el tratamiento de hemorragias en pacientes con hemofilia B con inhibidor.
FEIBA se puede utilizar para el tratamiento y la profilaxis de hemorragias en pacientes no hemofílicos con inhibidor adquirido del factor VIII.
Además, FEIBA se utiliza para la profilaxis en intervenciones quirúrgicas en pacientes con hemofilia A con inhibidor.
FEIBA se puede utilizar en todos los grupos de edad.

2. Qué necesita saber antes de empezar a usar FEIBA
Informe a su médico si tiene alguna alergia conocida.
Informe a su médico si está siguiendo una dieta pobre en sodio.
No use FEIBA
Solo se debe utilizar FEIBA en las siguientes circunstancias si, por ejemplo, debido a un título muy elevado de inhibidores no se espera ninguna respuesta al tratamiento con el concentrado de factor de la coagulación apropiado:
- si es alérgico (hipersensible) al complejo coagulante antiinhibidor o a alguno de los demás componentes de este medicamento (incluidos en la sección 6).
- si existe una coagulación intravascular diseminada (CID). (CID = coagulopatía de consumo, una condición potencialmente mortal en la que hay una coagulación excesiva de la sangre, con formación pronunciada de coágulos de sangre en los vasos sanguíneos. Esto provoca un consumo de los factores de coagulación en todo el organismo).
- en caso de infarto de miocardio, trombosis y/o embolia aguda: FEIBA debe utilizarse únicamente en los episodios hemorrágicos que constituyan una amenaza para la vida.
Advertencias y precauciones
Consulte a su médico antes de empezar a usar FEIBA porque pueden producirse reacciones de hipersensibilidad, como en el caso de todos los productos derivados del plasma que se administran por vía intravenosa. Para poder reconocer una reacción alérgica lo antes posible, debe conocer que los primeros síntomas potenciales de una reacción de hipersensibilidad pueden ser:
- eritema (enrojecimiento de la piel)
- erupción cutánea
- aparición de habones en la piel (urticaria)
- picor por todo el cuerpo
- hinchazón de los labios y de la lengua
- dificultad para respirar/disnea
- opresión en el pecho
- malestar general
- mareo
- caída de la tensión arterial
Otros síntomas de reacciones de hipersensibilidad a productos derivados del plasma incluyen letargia y cansancio.
Si nota alguno de estos síntomas, debe parar la administración inmediatamente y contactar con su médico de inmediato. Los síntomas descritos pueden indicar un shock anafiláctico. Los síntomas intensos requieren un tratamiento temprano de urgencia.
Su médico solamente reutilizará FEIBA en pacientes con sospecha de hipersensibilidad al producto o a alguno de sus componentes después de valorar detenidamente el beneficio esperado y el riesgo de la reexposición y/o de no esperar ninguna respuesta con otro tratamiento preventivo o terapia alternativa.
- Si experimenta cambios importantes en la tensión arterial o en la frecuencia del pulso, dificultades para respirar, tos o dolor en el pecho, debe parar la administración inmediatamente y contactar con su médico. Su médico iniciará las medidas de diagnóstico y terapéuticas apropiadas.
- En pacientes con hemofilia con inhibidores o con inhibidores adquiridos a los factores de coagulación. Durante el tratamiento con FEIBA, estos pacientes pueden tener un aumento en la tendencia a desarrollar hemorragia y un aumento del riesgo de trombosis al mismo tiempo.
Durante el tratamiento con FEIBA han aparecido acontecimientos trombóticos y tromboembólicos, incluyendo coagulación intravascular diseminada (CID), trombosis venosa, embolia pulmonar, infarto de miocardio e ictus. Es probable que el uso concomitante con factor VIIa recombinante aumente el riesgo de desarrollar un acontecimiento tromboembólico. Algunos de los acontecimientos tromboembólicos han ocurrido con el tratamiento de altas dosis de FEIBA.
En un ensayo realizado por otra compañía para evaluar emicizumab (un medicamento para prevenir sangrados en pacientes con hemofilia A), algunos pacientes que sufrieron sangrados intercurrentes fueron tratados con FEIBA para controlar los sangrados y, algunos de estos pacientes, desarrollaron microangiopatía trombótica (MAT). MAT es una condición grave y potencialmente amenazante para la vida. Cuando se tiene esta condición, se puede dañar la pared vascular y se pueden desarrollar coágulos en los vasos sanguíneos pequeños. En algunos casos, esto puede ocasionar daño de los riñones y otros órganos. En caso de sangrados intercurrentes mientras se está en profilaxis con emicizumab, contacte inmediatamente con su hematólogo o con su Centro de Tratamiento de Hemofilia.
Cuando se administran medicamentos derivados de plasma o sangre humano, hay que llevar a cabo ciertas medidas para evitar que las infecciones pasen a los pacientes. Tales medidas incluyen una cuidadosa selección de los donantes, para excluir a aquellos que están en riesgo de ser portadores de enfermedades infecciosas, análisis de marcadores específicos de infecciones en las donaciones individuales y en las mezclas de plasma, así como la inclusión de etapas en el proceso de fabricación para eliminar/inactivar virus. A pesar de esto, cuando se administran medicamentos derivados de la sangre o plasma humanos, la posibilidad de transmisión de agentes infecciosos no se puede excluir totalmente. Esto también se refiere a virus emergentes o de naturaleza desconocida u otros tipos de infecciones.
Estas medidas se consideran eficaces para virus envueltos como el virus de la inmunodeficiencia humana (VIH), virus de la hepatitis B y virus de la hepatitis C y para los virus no envueltos de la hepatitis A. Las medidas adoptadas pueden tener un valor limitado para virus no envueltos tales como el parvovirus B19. La infección por parvovirus B19 puede ser grave para una mujer embarazada (infección fetal) y para individuos cuyo sistema inmunitario está deprimido o para pacientes con algún tipo de anemia (por ejemplo enfermedad drepanocítica o anemia hemolítica).
Es posible que su médico le recomiende vacunarse frente a hepatitis A y hepatitis B, si a usted se le administra de forma regular o repetida productos derivados del plasma para los inhibidores del factor VIII.
Después de la administración de dosis elevadas de FEIBA, el aumento transitorio de los anticuerpos de superficie de la hepatitis B transferidos pasivamente puede provocar una interpretación errónea de los resultados positivos del test serológico.
FEIBA es un derivado de plasma y puede contener sustancias que reaccionan cuando se perfunden a los pacientes, causando la presencia de isohemaglutininas (anticuerpos que provocan la adhesión de los glóbulos rojos de otra persona). Este proceso puede llevar a malinterpretar los resultados de los análisis de sangre.
Se recomienda encarecidamente que cada vez que se administre una dosis de FEIBA, se deje constancia del nombre del medicamento y número de lote administrado con el fin de mantener un registro de los lotes utilizados.
Niños
La experiencia en niños menores de 6 años es limitada; debe adaptarse el mismo régimen posológico que en los adultos para el estado clínico del niño.
Otros medicamentos y FEIBA
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o pudiera tener que utilizar cualquier otro medicamento.
No se han realizado estudios adecuados y bien controlados del uso combinado o secuencial de FEIBA y factor VIIa recombinante, antifibrinolíticos o emicizumab. Cuando se usan antifibrinolíticos sistémicos, como ácido tranexámico y ácido aminocaproico, durante el tratamiento con FEIBA se debe considerar la posibilidad de aparición de acontecimientos tromboembólicos. Por lo tanto, no se deben utilizar antifibrinolíticos hasta aproximadamente 6 a 12 horas después de la administración de FEIBA.
De acuerdo a los datos in vitro disponibles y a las observaciones clínicas, no se puede excluir una interacción potencial medicamentosa con el uso concomitante con factor VIIa recombinante que potencialmente produzca un acontecimiento tromboembólico. Informe a su médico si va a ser tratado con FEIBA después de haber recibido emicizumab (un medicamento para prevenir los sangrados en pacientes con hemofilia A) ya que hay que tener en cuenta algunas advertencias y precauciones específicas. Su médico necesitará hacerle un seguimiento estrecho.
Como con todos los productos utilizados para la coagulación sanguínea, FEIBA no debe mezclarse con otros medicamentos antes de su administración ya que esto puede perjudicar a la eficacia y tolerancia del producto. Es conveniente lavar la vía venosa con solución salina isotónica antes y después de la administración de FEIBA.
Embarazo, lactancia y fertilidad
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de utilizar este medicamento.
Su médico decidirá si FEIBA puede ser usado durante el embarazo y la lactancia. Debido al aumento del riesgo de trombosis durante el embarazo, FEIBA solo debe ser administrado bajo una estrecha supervisión médica y solo si se encuentra claramente indicado. Para información sobre el riesgo de infección por parvovirus B19, ver sección advertencias y precauciones.
Conducción y uso de máquinas
No hay ningún signo de que FEIBA pueda afectar a la capacidad para conducir y utilizar máquinas.
FEIBA contiene sodio
500 U
Este medicamento contiene aproximadamente 40 mg de sodio (componente principal de la sal de mesa/para cocinar) en cada vial. Esto equivale al 2 % de la ingesta diaria máxima de sodio recomendada para un adulto.
1 000 U
Este medicamento contiene aproximadamente 80 mg de sodio (componente principal de la sal de mesa/para cocinar) en cada vial. Esto equivale al 4 % de la ingesta diaria máxima de sodio recomendada para un adulto.
2 500 U
Este medicamento contiene aproximadamente 200 mg de sodio (componente principal de la sal de mesa/para cocinar) en cada vial. Esto equivale al 10 % de la ingesta diaria máxima de sodio recomendada para un adulto.
3. Cómo usar FEIBA
Reconstituir el polvo liofilizado de FEIBA con el disolvente incluido y administre la solución por vía intravenosa.
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico o farmacéutico.
Su médico determinará la frecuencia y la dosis requerida para usted personalmente teniendo en cuenta la gravedad del trastorno de la coagulación sanguínea, la localización y la magnitud de la hemorragia, y el estado clínico y la respuesta a la preparación. No cambie la dosificación establecida por su médico y no suspenda la administración del preparado.
Si tiene la impresión de que el efecto de FEIBA es demasiado fuerte o demasiado débil, consulte a su médico o farmacéutico.
Calentar el producto a temperatura ambiente o a temperatura corporal antes de su administración, si es necesario.
FEIBA debe reconstituirse inmediatamente antes de su administración. La solución debe utilizarse de inmediato (ya que la preparación no contiene conservantes).
Agitar suavemente hasta que todo el producto esté disuelto. Asegurar que FEIBA esté completamente disuelto ya que, de lo contrario, pasarán menos unidades de FEIBA a través del filtro del equipo.
Las soluciones de aspecto turbio o que contengan depósitos se deben eliminar adecuadamente.
No reutilizar los envases abiertos.
Utilizar únicamente el agua para preparaciones inyectables y el equipo para la reconstitución incluidos en el envase.
Si se utilizan otros equipos distintos a los incluidos, asegúrese de usar un filtro adecuado de, al menos, 149 micrómetros de tamaño de poro.
No utilizar el producto si su sistema de barrera de esterilidad o su envase están dañados o muestran cualquier signo de deterioro.
No refrigerar después de la reconstitución.
Tras la completa reconstitución de FEIBA, la inyección o perfusión debe comenzar inmediatamente y debe completarse en el plazo de 3 horas desde la reconstitución.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo a la normativa local.
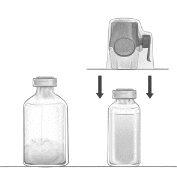
Reconstitución del polvo para la preparación de una solución para perfusión con el equipo BAXJECT II Hi-Flow:
- Calentar el vial de disolvente sin abrir (agua para preparaciones inyectables) a temperatura ambiente o máximo 37°C, si es necesario, por ejemplo, utilizando un baño de agua durante varios minutos.
- Quitar los tapones protectores del vial de polvo y del vial de disolvente, y desinfectar los tapones de goma de ambos viales. Colocar los viales en una superficie lisa.
- Abrir el envoltorio del equipo BAXJECT II Hi-Flow quitando la lámina protectora sin tocar el contenido del envase (Figura a). No sacar el equipo del envoltorio.
- Dar la vuelta al envoltorio e insertar la punta de plástico transparente a través del tapón de goma del vial de disolvente (Figura b). Sacar ahora el equipo BAXJECT II Hi-Flow de su envoltorio (Figura c). No quite el tapón protector azul del equipo BAXJECT II Hi-Flow.
- Con el equipo BAXJECT II Hi-Flow unido al vial de disolvente, invertir el sistema de tal forma que el vial de disolvente esté en la parte superior del equipo. Insertar la punta de plástico de color púrpura del equipo BAXJECT II Hi-Flow a través del tapón del vial de FEIBA. El vacío hará que el disolvente penetre en el vial de FEIBA (Figura d).
- Agitar con suavidad, sin sacudir, todo el sistema hasta que todo el polvo se haya disuelto. Asegúrese de que FEIBA esté completamente disuelto, de otra manera el material activo puede quedar retenido en el filtro del equipo.
Figura a | Figura b | Figura c |
|
|
|


Perfusión
¡Usar una técnica aséptica durante todo el procedimiento!
- Quitar el tapón protector azul del equipo BAXJECT II Hi-Flow. Conectar firmemente la jeringa al equipo BAXJECT II Hi-Flow. NO INTRODUCIR AIRE EN LA JERINGA. (Figura e). Se recomienda encarecidamente utilizar una jeringa Luer Lock con objeto de asegurar una conexión firme entre la jeringa y el equipo BAXJECT II Hi-Flow (girar la jeringa en el sentido de las agujas del reloj hasta que se pare cuando llegue al tope).
- Invertir el sistema para que el producto disuelto se encuentre en la parte de arriba. Introducir el producto disuelto en la jeringa, tirando del émbolo hacia atrás LENTAMENTE y asegurar que la conexión firme entre el dispositivo BAXJECT II Hi-Flow y la jeringa se mantiene durante todo el proceso mientras se tira del émbolo de la jeringa (Figura f).
- Desconectar la jeringa.
- Si se produce espuma dentro de la jeringa, esperar a que la espuma se compacte. Administrar lentamente por vía intravenosa la solución con el equipo de perfusión suministrado.
Figura d | Figura e | Figura f |
|
|
|

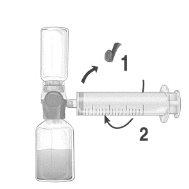

No exceder una velocidad de perfusión de 10U de FEIBA/kg por minuto.
Si usa más FEIBA del que debe
Informe inmediatamente a su médico. La sobredosis de FEIBA puede aumentar el riesgo de efectos adversos, como tromboembolia (formación de un coágulo de sangre con enrojecimiento en los vasos sanguíneos), coagulación intravascular diseminada (CID) o infarto de miocardio. Algunos de los acontecimientos tromboembólicos notificados se produjeron con dosis superiores a 200 U/kg/día o con pacientes con otros factores de riesgo de sufrir acontecimientos tromboembólicos. Si se observan signos o síntomas de acontecimiento tromboembólico, la perfusión se debe interrumpir inmediatamente y se deben adoptar las medidas de diagnóstico y terapéuticas necesarias.
En caso de sobredosis o ingestión accidental, consulte inmediatamente a su médico o farmacéutico o llame al Servicio de Información Toxicológica, teléfono 91 562 04 20, indicando el medicamento y la cantidad ingerida.

4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Efectos adversos frecuentes(pueden afectar hasta 1 de cada 10 pacientes)
Hipersensibilidad, dolor de cabeza, mareo, hipotensión, erupción cutánea, anticuerpos de superficie de la hepatitis B positivos.
Efectos adversos con frecuencia desconocida(no puede estimarse a partir de los datos disponibles)
Trastornos de la sangre y del sistema linfático:coagulación intravascular diseminada (CID), aumento del título de inhibidor.
Trastornos del sistema inmunológico:reacciones anafilácticas, erupción cutánea por todo el cuerpo (urticaria).
Trastornos del sistema nervioso:sensación de entumecimiento de las extremidades (hipoestesia), sensibilidad anormal o reducida (parestesia), accidente cerebrovascular (accidente trombótico, accidente embólico), somnolencia, sensación del gusto alterada (disgeusia).
Trastornos cardíacos:ataque cardíaco (infarto de miocardio), palpitación del corazón (taquicardia).
Trastornos vasculares:formación de coágulos de sangre con enrojecimiento de los vasos sanguíneos (acontecimientos tromboembólicos, trombosis venosa y arterial), aumento de la tensión arterial (hipertensión), rubor.
Trastornos respiratorios, torácicos y mediastínicos:obstrucción de la arteria pulmonar (embolia pulmonar), obstrucción del paso del aire (broncoespasmo), pitidos en el pecho, tos, dificultad al respirar (disnea).
Trastornos gastrointestinales:vómitos, diarrea, malestar abdominal, sensación de enfermedad (náuseas).
Trastornos de la piel y del tejido subcutáneo:sensación de entumecimiento en la cara, hinchazón de la cara, de la lengua y de los labios (angioedema), erupción cutánea por todo el cuerpo (urticaria), picor (prurito).
Trastornos generales y alteraciones en el lugar de administración:dolor en el lugar de la inyección, malestar general, sensación de calor, escalofríos, fiebre, dolor en el pecho, malestar en el pecho.
Exploraciones complementarias:caída de la tensión arterial, aumento del nivel de dímero D de fibrina en sangre.
La perfusión intravenosa rápida puede causar un dolor punzante y una sensación de entumecimiento en la cara y en las extremidades, así como una disminución de la tensión arterial.
Se notificaron casos de infarto de miocardio después de la administración de dosis superiores a la dosis máxima diaria y/o con administraciones prolongadas y/ o de la presencia de factores de riesgo tromboembólicos.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano: www.notificaram.es.
Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de FEIBA
Mantener este medicamento fuera de la vista y del alcance de los niños.
No conservar a temperatura superior a 25 °C. No congelar.
Conservar en el embalaje original para protegerlo de la luz.
No utilice este medicamento después de la fecha de caducidad que aparece en la etiqueta y en el envase. La fecha de caducidad es el último día del mes que se indica.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional
Composición de FEIBA
Polvo
- El principio activo por vial es complejo coagulante antiinhibidor.
- 1 ml contiene 100 U de complejo coagulante antiinhibidor.
- FEIBA 100 U/ml está disponible en tres presentaciones:
- La presentación de 500 U de FEIBA contiene 500 U (unidades) de complejo coagulante antiinhibidor en 200 – 600 mg de proteína plasmática humana.
- La presentación de 1 000 U de FEIBA contiene 1 000 U (unidades) de complejo coagulante antiinhibidor en 400 – 1 200 mg de proteína plasmática humana.
- La presentación de 2 500 U de FEIBA contiene 2 500 U (unidades) de complejo coagulante antiinhibidor en 1 000 – 3 000 mg de proteína plasmática humana.
FEIBA contiene también los factores II, IX y X, principalmente no activados, así como factor VII activado. El antígeno del factor VIII coagulante (FVIII C:Ag) así como los factores del sistema de calicreína-cinina están presentes sólo en cantidades trazas.
- Los demás componentes son cloruro de sodio y citrato de sodio.
Disolvente
- Agua para preparaciones inyectables.
Aspecto del producto y contenido del envase
El producto se presenta como un polvo liofilizado o sólido friable, de color blanco a blanquecino o verde pálido. La solución reconstituida tiene un pH entre 6,5 y 7,3.
El polvo y el disolvente se suministran en viales de vidrio cerrados con tapones de goma.
Presentación:1 x 500 U
1 x 1 000 U
1 x 2 500 U
Puede que solamente estén comercializados algunos tamaños de envases.
Contenido del envase:
- 1 vial con 500 U/1 000 U/2 500 U de FEIBA - polvo para solución para perfusión
- 1 vial con 5 ml/10 ml/25 ml de agua para preparaciones inyectables
- 1 equipo para reconstitución BAXJECT II Hi-Flow
- 1 jeringa desechable
- 1 aguja mariposa
Titular de la autorización de comercialización y responsable de la fabricación
Titular de la autorización de comercialización:
Baxalta Innovations GmbH
Industriestrasse, 67
1221 Viena, Austria
Responsable de la fabricación:
Takeda Manufacturing Austria AG
Industriestrasse, 67
1221 Viena, Austria
Representante local del titular
Takeda Farmacéutica España, S.A.
Calle Albacete, 5, planta 9ª,
Edificio Los Cubos
28027 Madrid
España
Tel: +34 91 790 42 22
Este medicamento está autorizado en los Estados miembros del Espacio Económico Europeo con los siguientes nombres:
Austria:
FEIBA 100 E./ml Pulver und Lösungsmittel zur Herstellung einer Infusionslösung
Bulgaria:
FEIBA 100 U/ml powder and solvent for solution for infusion
Croacia:
FEIBA 100 U/ml prašak i otapalo za otopinu za infuziju
Chipre:
FEIBA 100 U/ml κ?νις και διαλ?της για δι?λυμα προς ?γχυση
República Checa:
FEIBA
Dinamarca:
Feiba
Estonia:
FEIBA 100 Ü/ML
Finlandia:
Feiba
Alemania:
FEIBA 500 E konzentriert
FEIBA 1000 E konzentriert
FEIBA 2500 E konzentriert
Grecia:
FEIBA 100 U/ml κ?νις και διαλ?της για δι?λυμα προς ?γχυση
Irlanda:
FEIBA 100 U/ml powder and solvent for solution for infusion
Letonia:
Feiba 100 V/ml pulveris un škidinatajs infuziju škiduma pagatavošanai
Lituania:
Feiba 100 V/ml milteliai ir tirpiklis infuziniam tirpalui
Malta:
FEIBA 100 U/ml powder and solvent for solution for infusion
Países Bajos:
FEIBA 100 E/ML, poeder en oplosmidel voor oplossing voor injectie
Noruega:
Feiba
Rumanía:
FEIBA 100 U/ml pulbere si solvent pentru solutie injectabila
Eslovaquia:
FEIBA 100 U/ml prášok a rozpúšt’adlo na infúzny roztok
Eslovenia:
FEIBA 100 e./ml prašek in vehikel za raztopino za infundiranje
España:
FEIBA 100 U/ml polvo y disolvente para solución para perfusión
Suecia:
Feiba 100 enheter/ml pulver och vätska till infusionsvätska, lösning
Fecha de la última revisión de este prospecto:08/2024
La información detallada de este medicamento está disponible en la página web de la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) http://www.aemps.gob.es/
---------------------------------------------------------------------------------------------------------------------
Esta información está destinada únicamente a profesionales del sector sanitario:
El tratamiento se debe iniciar y monitorizar bajo la supervisión de un médico con experiencia en el tratamiento de los trastornos de la coagulación.
Posología
La dosis y la duración de la terapia dependen de la gravedad de la alteración de la función hemostática, de la localización y la gravedad de la hemorragia y del estado clínico del paciente.
La dosis y la frecuencia de la administración se establecerán siempre en función de la eficacia clínica en cada caso.
Como guía general, se recomiendan dosis de 50 – 100 U/kg de FEIBA; no debe sobrepasarse una dosis única de 100 U/kg ni una dosis máxima diaria de 200 U/kg, a menos que la gravedad de la hemorragia requiera y justifique el uso de dosis superiores.
A causa de factores específicos de los pacientes, la respuesta a un agente de bypass puede variar, y en una situación de hemorragia determinada, los pacientes con una respuesta insuficiente a un agente pueden responder a otro agente. En caso de respuesta insuficiente a un agente de bypass, se deberá plantear el uso de otro agente.
Población pediátrica
La experiencia en niños menores de 6 años es limitada; debe adaptarse el mismo régimen posológico que en los adultos para el estado clínico del niño.
- Hemorragia espontánea
Hemorragias en articulaciones, músculos y tejido blando
Para hemorragias leves a moderadas se recomienda una dosis de 50 ?75 U/kg, cada 12 horas. El tratamiento debe continuar hasta que aparezcan claros signos de mejoría clínica, tales como disminución del dolor, reducción de la tumefacción o aumento de la movilización de la articulación.
Para hemorragias intensas en músculos y tejido blando, p. ej., hemorragia retroperitoneal, se recomienda una dosis de 100 U/kg cada 12 horas.
Hemorragias en membranas mucosas
Se recomienda una dosis de 50 U/kg cada 6 horas, bajo estricta vigilancia del paciente (control visual del sangrado, repetición del hematocrito). Si no se detiene la hemorragia, se puede aumentar la dosis a 100 U/kg; sin embargo, no debe sobrepasarse una dosis de 200 U/kg.
Otras hemorragias intensas
En hemorragia intensa, como hemorragia del SNC, se recomienda una dosis de 100 U/kg cada 12 horas. En casos particulares, se puede administrar FEIBA cada 6 horas, hasta que aparezcan claros signos de mejoría clínica (no debe sobrepasarse la dosis máxima diaria de 200 U/kg).
- Cirugía
En intervenciones quirúrgicas, puede administrarse una dosis inicial de 100 U/kg antes de la operación, y entre las 6 a 12 horas posteriores puede administrarse otra dosis de 50 – 100 U/kg. Como dosis de mantenimiento postoperatorio se pueden administrar 50 – 100 U/kg cada 6 – 12 horas; la dosis, los intervalos de dosificación y la duración del tratamiento peri- y postoperatorio dependen de la intervención quirúrgica, del estado general del paciente y de la eficacia clínica en cada caso particular (no debe sobrepasarse la dosis máxima diaria de 200 U/kg).
- Profilaxis en pacientes con hemofilia A coninhibidor
- Profilaxis de hemorragias en pacientes con un título alto de inhibidor y hemorragias frecuentes, después de una respuesta fallida frente a la inducción de la inmunotolerancia (ITI) o cuando dicha inducción no se considera:
Se recomienda una dosis de 70 ? 100 U/kg en días alternos. Si fuese necesario, la dosis puede aumentarse a 100 U/kg al día o puede reducirse gradualmente.
- Profilaxis de hemorragias en pacientes con un título alto de inhibidor durante la inducción de la inmunotolerancia (ITI):
Se puede administrar FEIBA en combinación con factor VIII, en un intervalo de dosis de 50 ? 100 U/kg, dos veces al día, hasta que el título del inhibidor del factor VIII haya disminuido a < 2 U.B.*
*1 Unidad Bethesda se define como la cantidad de anticuerpos que inhibe el 50 % de la actividad del factor VIII en plasma incubado (2 horas a 37 °C).
- Uso de FEIBA en grupos especiales de pacientes
FEIBA también se ha utilizado en combinación con un concentrado de factor VIII como tratamiento de larga duración para la eliminación completa y permanente del inhibidor del factor VIII.
Monitorización
En caso de una respuesta inadecuada al tratamiento con el producto, se recomienda realizar un recuento plaquetario, dado que se considera necesario un número suficiente de plaquetas funcionalmente intactas para que el tratamiento con el producto sea eficaz.
Debido al complejo mecanismo de acción, no se dispone de una monitorización directa de los principios activos. Las pruebas de coagulación como el tiempo de coagulación en sangre total (TCT), el tromboemblastograma (TEG, valor r) y el tiempo de tromboplastina parcial activada (TTPa), generalmente muestran sólo pequeños acortamientos, que no se correlacionan necesariamente con la mejoría clínica. Por estas razones la utilidad de estos ensayos para monitorizar el tratamiento con FEIBA, es muy limitada.
Forma de administración
FEIBA debe administrarse lentamente, por vía intravenosa. FEIBA se debe perfundir a una velocidad de perfusión de 2 U/kg/min. En pacientes que hayan tolerado bien la velocidad de perfusión de 2 U/kg/min, la velocidad de perfusión puede aumentarse hasta un máximo de 10 U/kg/min.
FEIBA debe reconstituirse inmediatamente antes de su administración. La solución debe utilizarse de inmediato (ya que la preparación no contiene conservantes). No utilice soluciones de aspecto turbio o que contengan depósitos. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo a la normativa local.
Monitorización de la terapia
No deben sobrepasarse dosis individuales de 100 U/kg ni dosis diarias de 200 U/kg. Los pacientes que reciben más de 100 U/kg deben ser monitorizados para el desarrollo de CID y/o isquemia coronaria aguda y para síntomas de acontecimientos trombóticos o tromboembólicos. Para detener una hemorragia deben administrarse dosis altas de FEIBA sólo durante el tiempo que sea estrictamente necesario.
Si se producen cambios clínicamente significativos en la tensión arterial o en la frecuencia del pulso, dificultad respiratoria, tos o dolor en el pecho, se debe interrumpir la perfusión inmediatamente y se deben adoptar las medidas de diagnóstico y terapéuticas necesarias. Los parámetros analíticos característicos de CID son descenso del fibrinógeno, descenso del recuento de trombocitos y/o presencia de productos de degradación de la fibrina o del fibrinógeno (PDF). Otros parámetros para el desarrollo de CID son una clara prolongación del tiempo de trombina, del tiempo de protrombina o del tiempo de tromboplastina parcial activada TTPa. En pacientes con hemofilia con inhibidor o con inhibidores adquiridos de los factores VIII, IX y/o XI, el TTPa se encuentra prolongado a causa de la enfermedad subyacente.
La administración de FEIBA en pacientes con inhibidor puede producir un incremento inicial “anamnésico” de los niveles del inhibidor. Durante la administración continuada de FEIBA, los niveles de inhibidores pueden descender a lo largo del tiempo. Tanto los datos clínicos como los datos publicados sugieren que la eficacia de FEIBA no se reduce.
Los pacientes con hemofilia con inhibidor o con inhibidores adquiridos de factores de la coagulación que reciben tratamiento con FEIBA pueden ser más propensos a padecer hemorragia al tiempo que puede aumentar el riesgo de trombosis.
Pruebas de laboratorio y eficacia clínica
Los ensayos in vitro para controlar la eficacia, tales como TTPa, tiempo de coagulación total (TCT) y tromboelastograma (TEG) no tienen necesariamente una correlación con la mejora clínica. Por esta razón, no se debe buscar la normalización de estos valores mediante un incremento de dosis de FEIBA e incluso son fuertemente rechazados debido al posible riesgo de aparición de CID por sobredosis.
Importancia del recuento plaquetario
En caso de respuesta inadecuada al tratamiento con FEIBA, se recomienda realizar un recuento plaquetario, dado que se considera necesario un número suficiente de plaquetas funcionalmente intactas para que el tratamiento con FEIBA sea eficaz.
Tratamiento de pacientes con hemofilia B con inhibidor
La experiencia en pacientes con hemofilia B con inhibidor del factor IX es limitada debido a la rareza de la enfermedad. Cinco pacientes con hemofilia B con inhibidor fueron tratados con FEIBA durante los ensayos clínicos realizados, o con tratamiento a demanda o con tratamiento profiláctico o por intervenciones quirúrgicas:
En un estudio clínico prospectivo, abierto, aleatorizado y paralelo en pacientes con hemofilia A o B con título de inhibidor constantemente elevado (090701, PROOF), se aleatorizó a 36 pacientes para recibir tratamiento profiláctico o a demanda, durante 12 meses± 14 días. Los 17 pacientes del grupo con tratamiento profiláctico recibieron 85 ± 15 U/kg de FEIBA, administrado a días alternos y los 19 pacientes del grupo con tratamiento a demanda recibieron tratamiento individual, determinado por el médico. Dos pacientes con hemofilia B con inhibidor recibieron tratamiento a demanda y un paciente con hemofilia B recibió tratamiento profiláctico. La mediana de la Tasa Anual de Hemorragias (TAH) para todos los tipos de episodios hemorrágicos en los pacientes del grupo con tratamiento profiláctico (mediana de la TAH = 7,9) fue inferior a la de los pacientes del grupo con tratamiento a demanda (mediana de la TAH = 28,7) lo que supone una disminución del 72,5% de la mediana de la TAH entre los grupos de tratamiento.
En otro estudio prospectivo finalizado, de vigilancia de seguridad o no intervencionista del uso perioperatorio de FEIBA (PASS-INT-003, SURF), se realizaron un total de 34 intervenciones quirúrgicas en 23 pacientes. La mayoría de los pacientes (18) padecían hemofilia A congénita con inhibidor, dos eran pacientes con hemofilia B con inhibidor y tres eran pacientes con hemofilia A adquirida con inhibidor. El tiempo de exposición a FEIBA varió entre 1 y 28 días, con una media de 9 días y una mediana de 8 días. La dosis media acumulada fue de 88 347 U y la mediana de la dosis fue de 59 000 U. En los pacientes con hemofilia B con inhibidor, la exposición más prolongada a FEIBA fue de 21 días, y la dosis máxima administrada fue de 7 324 U.
También se han descrito en la bibliografía 48 pacientes en los que FEIBA se utilizó para el tratamiento y la prevención de episodios hemorrágicos en pacientes con hemofilia B con inhibidor del factor IX (34 pacientes con hemofilia B con inhibidor recibieron tratamiento a demanda, seis pacientes con hemofilia B con inhibidor recibieron tratamiento profiláctico y ocho pacientes con hemofilia B con inhibidor recibieron tratamiento por intervenciones quirúrgicas).
En un estudio cruzado prospectivo, abierto y aleatorizado (091501) se estudiaron la tolerabilidad y la seguridad de FEIBA reconstituido con un volumen regular o reducido en un 50%, así como con velocidades de perfusión más rápidas, en pacientes con hemofilia con inhibidor. Se trató a 33 pacientes y 28 pacientes completaron el estudio. Durante el estudio, se reconstituyó FEIBA con un volumen reducido en un 50% (concentración de 100 U/ml) y se perfundió por vía intravenosa a velocidades de 2, 4 y 10 U/kg/min a la dosis indicada de 85 U/kg ± 15 U/kg para todos los pacientes. Los criterios de valoración primarios fueron la tolerabilidad y la seguridad con el volumen reducido al 50% (mayor concentración) y con las velocidades de perfusión estándar y aumentadas. En el estudio se demostró que tanto la mayor concentración (100 U/ml) como las velocidades de perfusión aumentadas (4 y 10 U/kg/min) fueron bien toleradas y que el perfil de seguridad era comparable al administrar la dosis indicada de 85 U/kg ± 15 U/kg. Los pacientes que recibieron el volumen reducido al 50% (mayor concentración) a la velocidad de perfusión estándar de 2 U/kg/min presentaron tasas similares de acontecimientos adversos derivados del tratamiento (AADT) relacionados que los pacientes que recibieron la dosis con el volumen habitual (concentración de 50 U/ml) y con la misma velocidad de perfusión. Con la velocidad de perfusión de 4 U/kg/min no se notificaron AADT relacionados. Los pacientes que recibieron el volumen reducido al 50% (100 U/ml) a la velocidad de perfusión de 10 U/kg/min experimentaron 1 AADT relacionado que no fue grave. Además, los pacientes que recibieron el volumen reducido al 50% (concentración aumentada) a las velocidades de perfusión de 4 y 10 U/kg/min no experimentaron ningún AADT grave, reacciones de hipersensibilidad, reacciones en el lugar de perfusión, AADT trombótico ni AADT que conllevaran la interrupción del fármaco ni el abandono del estudio. En general, los AADT observados en el estudio fueron coherentes con el perfil de seguridad conocido de FEIBA en pacientes con hemofilia con inhibidor.
En un estudio de seguridad observacional, posautorización, abierto, no controlado y no intervencionista de FEIBA (PASS-EU-006), se trató con FEIBA a 75 pacientes (mediana de edad de 34,8 años, 70 hombres y 5 mujeres), de los cuales 73 tenían hemofilia A con inhibidor o hemofilia B con inhibidor. De los 65 pacientes que tenían hemofilia congénita, 63 tenían hemofilia A congénita y 2 tenían hemofilia B congénita. En el inicio, a 43 pacientes se les prescribió FEIBA como tratamiento profiláctico y a 32 pacientes se les prescribió como tratamiento a demanda. Se emplearon velocidades de perfusión mayores (> 2 U/kg/min) en 6 pacientes pediátricos con edades comprendidas entre los 11 meses y los 11 años, y en 5 pacientes adolescentes con edades comprendidas entre los 13 y los 16 años. De las 320 perfusiones realizadas a una velocidad de perfusión disponible en 7 pacientes pediátricos y en 6 pacientes adolescentes, hubo 129 perfusiones (40,3%) en 2 pacientes (ambos pediátricos) con una velocidad de perfusión > 10 U/kg/min, 26 perfusiones (8,1%) en 7 pacientes (4 pediátricos; 3 adolescentes) con velocidades de perfusión > 4 y ≤ 10 U/kg/min, 135 perfusiones (42,2%) en 7 pacientes (3 pediátricos; 4 adolescentes) con una velocidad de perfusión > 2 y ≤ 4 U/kg/min, y 30 perfusiones (9,4%) en 3 pacientes (1 pediátrico; 2 adolescentes) con una velocidad de perfusión ≤ 2 U/kg/min.
Además se dispone de notificaciones aisladas sobre el uso de FEIBA en el tratamiento de pacientes con inhibidores adquiridos frente a los factores IX, X, XI y XIII.
En casos raros, FEIBA también se utilizó en pacientes con presencia de inhibidor del factor de von Willebrand.
- País de registro
- Principio activo
- Requiere recetaSí
- Fabricante
- Esta información es de carácter general y no sustituye la consulta con un profesional sanitario.
- Alternativas a FEIBA 100 U/ML POLVO Y DISOLVENTE PARA SOLUCIÓN PARA PERFUSIÓNForma farmacéutica: INYECTABLE PERFUSION, 50 U/mlPrincipio activo: factor VIII inhibitor bypassing activityFabricante: Baxalta Innovations GmbhRequiere recetaForma farmacéutica: INYECTABLE, 1.000 UIPrincipio activo: factor de coagulación VIIIFabricante: Takeda Manufacturing Austria AgRequiere recetaForma farmacéutica: INYECTABLE, 1.500 UIPrincipio activo: factor de coagulación VIIIFabricante: Takeda Manufacturing Austria AgRequiere receta
Médicos online para FEIBA 100 U/ML POLVO Y DISOLVENTE PARA SOLUCIÓN PARA PERFUSIÓN
Comenta la dosis, los posibles efectos secundarios, interacciones, contraindicaciones o la revisión de receta de FEIBA 100 U/ML POLVO Y DISOLVENTE PARA SOLUCIÓN PARA PERFUSIÓN, sujeto a valoración médica y a la normativa local.










Preguntas frecuentes
