

AFSTYLA 1.500 UI POLVO Y DISOLVENTE PARA SOLUCION INYECTABLE



Cómo usar AFSTYLA 1.500 UI POLVO Y DISOLVENTE PARA SOLUCION INYECTABLE
Traducción generada por IA
Este contenido ha sido traducido automáticamente y se ofrece solo con fines informativos. No sustituye la consulta con un profesional sanitario.
Ver originalContenido del prospecto
Introducción
Prospecto: información para el usuario
AFSTYLA 250 UI, polvo y disolvente para solución inyectable
AFSTYLA 500 UI, polvo y disolvente para solución inyectable
AFSTYLA 1.000 UI, polvo y disolvente para solución inyectable
AFSTYLA 1.500 UI, polvo y disolvente para solución inyectable
AFSTYLA 2.000 UI, polvo y disolvente para solución inyectable
AFSTYLA 2.500 UI, polvo y disolvente para solución inyectable
AFSTYLA 3.000 UI, polvo y disolvente para solución inyectable
lonoctocog alfa (factor VIII de coagulación recombinante de cadena única)
Lea todo el prospecto detenidamente antes de que usted o su hijo empiece a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted o a su hijo, y no debe dárselo a otras personas,aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
- Qué es AFSTYLA y para qué se utiliza
- Qué necesita saber antes de que usted o su hijo empiece a usar AFSTYLA
- Cómo usar AFSTYLA
- Posibles efectos adversos
- Conservación de AFSTYLA
- Contenido del envase e información adicional
1. Qué es AFSTYLA y para qué se utiliza
AFSTYLA es un producto con factor VIII de coagulación humano que se produce mediante tecnología de ADN recombinante. El principio activo de AFSTYLA es lonoctocog alfa.
AFSTYLA se usa para tratar y prevenir los episodios hemorrágicos en pacientes con hemofilia A (deficiencia congénita del factor VIII). El factor VIII es una proteína necesaria para la coagulación sanguínea. A los pacientes con hemofilia A les falta este factor, por lo cual la sangre no se coagula tan rápidamente como debería y hay una mayor tendencia a sangrar. AFSTYLA actúa reemplazando al factor VIII ausente en los pacientes con hemofilia A haciendo que su sangre pueda coagularse normalmente.
AFSTYLA se puede utilizar en todos los grupos de edad.

2. Qué necesita saber antes de empezar a usar AFSTYLA
No use AFSTYLA
- Si el paciente de Afstyla ha experimentado una reacción alérgica potencialmente mortal a AFSTYLA o a alguno de sus componentes (incluidos en la sección 6).
- Si el paciente de Afstyla es alérgico a las proteínas de hámster.
Advertencias y precauciones
Trazabilidad
Es importante llevar un registro del número de lote de AFSTYLA.
Por lo tanto, cada vez que use un nuevo paquete de AFSTYLA, anote la fecha y el número de lote (que está en la caja después de “Lote”) y guarde esta información en un lugar seguro.
Consulte a su médico, farmacéutico o enfermero antes de empezar a usar AFSTYLA.
- Es posible que se produzcan reacciones alérgicas (hipersensibilidad). El producto contiene restos de proteínas de hámster (ver también "No use AFSTYLA"). Si se manifiestan síntomas de alergia, interrumpa el tratamiento inmediatamente y contacte con su médico.Su médico debe informarle de los primeros signos de las reacciones de alergia. Estos incluyen ronchas, erupción cutánea generalizada, presión en el pecho, dificultad para respirar, caída de la presión arterial y anafilaxia (una reacción alérgica grave que causa dificultades respiratorias graves y mareos).
- La formación de inhibidores(anticuerpos) es una complicación conocida que puede producirse durante el tratamiento con todos los medicamentos compuestos por factor VIII. Estos inhibidores, especialmente en grandes cantidades, impiden que el tratamiento funcione correctamente. Usted o su hijo serán supervisados cuidadosamente por si desarrollan inhibidores. Si su hemorragia o la de su hijo no se está controlando con AFSTYLA, consulte a su médico inmediatamente.
- Si le han dicho que usted o su hijo padecen una enfermedad cardiaca o tiene riesgo de padecerla, informe a su médico o farmacéutico.
- Si se utiliza un dispositivo de acceso venoso central (DAVC) para la inyección de AFSTYLA, su médico debe considerar y comentarle el riesgo de complicaciones, como infecciones locales, bacterias en sangre (bacteriemia) y la formación de coágulos (trombosis) en los vasos sanguíneos en el lugar de inserción.
Otros medicamentos y AFSTYLA
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
Embarazo y lactancia
- Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de utilizar este medicamento.
- Durante el embarazo y el periodo de lactancia, AFSTYLA solo se debe administrar si es claramente necesario.
Conducción y uso de máquinas
AFSTYLA no afecta a su capacidad para conducir o usar máquinas.
AFSTYLA contiene sodio
Este medicamento contiene 35 mg de sodio (componente principal de la sal de mesa/para cocinar) en cada vial. Esto equivale al 1,8% de la ingesta diaria máxima de sodio recomendada para un adulto.
3. Cómo usar AFSTYLA
Su tratamiento debe ser supervisado por un médico con experiencia en el tratamiento de trastornos de coagulación de la sangre.
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico.
Dosis
La cantidad de AFSTYLA que usted o su hijo necesitan y la duración del tratamiento dependen:
- de la gravedad de su enfermedad
- del lugar y de la intensidad de la hemorragia
- de su estado clínico y su respuesta clínica
- de su peso corporal
Siga las instrucciones indicadas por su médico.
Reconstitución y administración
Instrucciones generales
- El polvo se debe mezclar con el disolvente (líquido) y extraerse del vial en condiciones asépticas.
- AFSTYLA no se debe mezclar con otros medicamentos o disolventes, excepto los mencionados en la sección 6.
- La solución debe ser transparente o ligeramente opalescente, entre amarilla e incolora, es decir, puede brillar cuando se expone a la luz pero no debe contener ninguna partícula visible. Después de filtrar o extraer la solución (véase más adelante) se debe volver a revisar antes de su uso. No utilice la solución si está visiblemente turbia o si contiene flóculos o partículas.
- La eliminación del producto no utilizado y de todos los materiales residuales se realizará de acuerdo con la normativa local y las indicaciones de su médico.
Reconstitución y administración
Sin abrir ninguno de los viales, asegúrese de que el polvo de AFSTYLA y el líquido estén a temperatura ambiente o corporal. Esto se puede hacer dejando los viales a temperatura ambiente durante una hora aproximadamente o sujetándolos con las manos durante unos minutos. No exponga los viales al calor directo. Los viales no deben calentarse por encima de la temperatura corporal (37 ºC).
Retire con cuidado las cápsulas protectoras de los viales y, a continuación, limpie la parte al descubierto de los tapones de goma con una toallita impregnada de alcohol. Deje secar los viales antes de abrir el envase del Mix2Vial (el cual contiene el trasvasador con filtro) y, a continuación, siga las instrucciones que se indican a continuación.
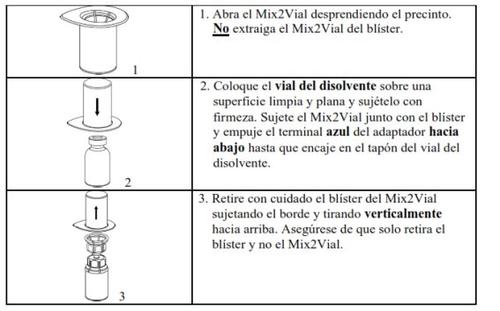

Utilice el kit de venopunción suministrado con el producto e inserte la aguja en una vena. Deje que la sangre fluya hasta el final del tubo. Acople la jeringa al extremo de bloqueo roscado del kit de venopunción. Inyecte lentamente la solución reconstituida (a una velocidad que le resulte cómoda, hasta un máximo de 10 ml/min) en la venasegún las instrucciones que le haya dado su médico. Intente que no entre sangre en la jeringa que contiene el producto.
Compruebe si experimenta efectos adversos justo después de la inyección. Si experimenta algún efecto adverso que pueda estar relacionado con la administración de AFSTYLA, la inyección debe interrumpirse (ver también la sección 2).
Uso en niños y adolescentes
AFSTYLA puede utilizarse en niños y adolescentes de todas las edades. En el caso de los niños menores de 12 años, puede que se necesiten dosis más altas o inyecciones más frecuentes. En niños mayores de 12 años, se puede utilizar la misma dosis que en adultos.
Si usa más AFSTYLA del que debe
Si se ha inyectado más AFSTYLA del que debe, informe de ello a su médico.
Si olvidó usar AFSTYLA
No se administre una dosis doble para compensar la dosis olvidada. Adminístrese inmediatamente la siguiente dosis y siga las instrucciones de su médico.
Si interrumpe el tratamiento con AFSTYLA
Si interrumpe el uso de AFSTYLA, puede dejar de estar protegido frente al sangrado o puede que no deje de sangrar si padece un sangrado actualmente. No deje de usar AFSTYLA sin consultarlo antes con su médico.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.

4. Posibles efectos adversos
Al igual que todos los medicamentos, AFSTYLA puede producir efectos adversos, aunque no todas las personas los sufran.
Deje de usar inmediatamente el medicamento y póngase en contacto con su médico:
- si nota síntomas de reacciones alérgicas
- Es posible que se produzcan reacciones alérgicas que incluyan los síntomas siguientes: ronchas, urticaria generalizada (erupción con picor), opresión torácica, sibilancia, presión sanguínea baja y anafilaxia (reacción grave que causa dificultad severa para respirar o mareo). Si esto sucede, debe interrumpir el medicamento inmediatamente y ponerse en contacto con su médico.
- si nota que el medicamento ha dejado de funcionar correctamente(sangrado no cesa) Para los niños que no han sido tratados previamente con medicamentos del factor VIII, los anticuerpos inhibidores (ver sección 2) pueden formarse con mucha frecuencia (más de 1 de cada 10 pacientes); sin embargo, en pacientes que han recibido tratamiento previo con factor VIII (más de 150 días de tratamiento) el riesgo es poco común (menos de 1 de cada 100 pacientes). Si usted o su hijo ha desarrollado un inhibidor debido a la medicina pueden experimentar un sangrado persistente. Si esto sucede, debe comunicarse con su médico inmediatamente.
Efectos adversos frecuentes (pueden afectar hasta 1 de cada 10 personas)
- Hormigueo o entumecimiento (parestesia).
- Erupción cutánea.
- Fiebre.
Efectos adversos poco frecuentes (pueden afectar hasta 1 de cada 100 personas)
- Prurito.
- Enrojecimiento de la piel.
- Dolor en la zona de inyección.
- Escalofríos.
- Sensación de calor.
Efectos adversos en niños y adolescentes
No se han observado diferencias específicas de la edad en las reacciones adversas entre los niños, los adolescentes y los adultos.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de AFSTYLA
- Mantener este medicamento fuera de la vista y del alcance de los niños.
- No utilice este medicamento después de la fecha de caducidad que aparece en la etiqueta y en la caja.
- Conservar en nevera (entre 2 °C y 8 °C).
- Antes de reconstituir el polvo de AFSTYLA, se puede conservar a temperatura ambiente (por debajo de los 25 °C) durante un período único no superior a los 3 meses, dentro de la fecha de caducidad impresa en las cajas y los viales. Anote la fecha en que empiece a conservar AFSTYLA a temperatura ambiente en la caja del medicamento.
- Una vez se ha sacado el medicamento de la nevera, no debe volver a meterlo en la misma.
- No congelar.
- Conserve el vial dentro de su caja para protegerlo de la luz.
- Una vez reconstituido el medicamento se debe utilizar preferentemente de inmediato.
- Si el producto reconstituido no se administra inmediatamente, los tiempos y las condiciones de conservación antes de su uso son responsabilidad del usuario.
6. Contenido del envase e información adicional
Composición de AFSTYLA
El principio activo es:
250 UI por vial; tras la reconstitución con 2,5 ml de agua para preparaciones inyectables, la solución contiene 100 UI/ml de lonoctocog alfa.
500 UI por vial; tras la reconstitución con 2,5 ml de agua para preparaciones inyectables, la solución contiene 200 UI/ml de lonoctocog alfa.
1.000 UI por vial; tras la reconstitución con 2,5 ml de agua para preparaciones inyectables, la solución contiene 400 UI/ml de lonoctocog alfa.
1.500 UI por vial; tras la reconstitución con 5 ml de agua para preparaciones inyectables, la solución contiene 300 UI/ml de lonoctocog alfa.
2.000 UI por vial; tras la reconstitución con 5 ml de agua para preparaciones inyectables, la solución contiene 400 UI/ml de lonoctocog alfa.
2.500 UI por vial; tras la reconstitución con 5 ml de agua para preparaciones inyectables, la solución contiene 500 UI/ml de lonoctocog alfa.
3.000 UI por vial; tras la reconstitución con 5 ml de agua para preparaciones inyectables, la solución contiene 600 UI/ml de lonoctocog alfa.
Los demás componentes son:
L-histidina, polisorbato 80, cloruro de calcio dihidratado, cloruro sódico (véase el último apartado de la sección 2), sacarosa.
Disolvente: agua para preparaciones inyectables.
Aspecto de AFSTYLA y contenido del envase
AFSTYLA se presenta en forma de polvo o masa friable de color blanco o ligeramente amarillento y disolvente para solución inyectable transparente e incoloro.
La solución reconstituida debe ser transparente o ligeramente opalescente, entre amarilla e incolora, es decir, puede brillar cuando se expone a la luz pero no debe contener ninguna partícula visible.
Presentaciones
Un envase con 250, 500 o 1.000 UI que contiene:
1 vial con polvo
1 vial con 2,5 ml de agua para preparaciones inyectables
1 trasvasador con filtro 20/20
Una caja interior que contiene:
1 jeringa de 5 ml desechable
1 equipo de venopunción
2 toallitas impregnadas de alcohol
1 apósito no estéril
Un envase con 1.500, 2.000, 2.500 o 3.000 UI que contiene:
1 vial con polvo
1 vial con 5 ml de agua para preparaciones inyectables
1 trasvasador con filtro 20/20
Una caja interior que contiene:
1 jeringa de 10 ml desechable
1 equipo de venopunción
2 toallitas impregnadas de alcohol
1 apósito no estéril
Puede que solamente estén comercializados algunos tamaños de envase.
Acondicionamientos primarios
250 UI | Vial de vidrio con tapón de goma, disco naranja de plástico y cáspsula de aluminio verde a rayas |
500 UI | Vial de vidrio con tapón de goma, disco azul de plástico y cáspsula de aluminio verde a rayas |
1.000 UI | Vial de vidrio con tapón de goma, disco verde de plástico y cáspsula de aluminio verde a rayas |
1.500 UI | Vial de vidrio con tapón de goma, disco turquesa de plástico y cáspsula de aluminio verde a rayas |
2.000 UI | Vial de vidrio con tapón de goma, disco púrpura de plástico y cáspsula de aluminio verde a rayas |
2.500 UI | Vial de vidrio con tapón de goma, disco gris claro de plástico y cáspsula de aluminio verde a rayas |
3.000 UI | Vial de vidrio con tapón de goma, disco amarillo de plástico y cáspsula de aluminio verde a rayas |
Titular de la autorización de comercialización y responsable de la fabricación
CSL Behring GmbH
Emil-von-Behring-Straße 76
35041 Marburg
Alemania
Puede solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
België/Belgique/Belgien CSL Behring NV Tél/Tel: +32 15 28 89 20 | Lietuva CentralPharma Communications UAB Tel: +370 5 243 0444 |
| Luxembourg/Luxemburg CSL Behring NV Tél/Tel: +32 15 28 89 20 |
Ceská republika CSL Behring s.r.o. Tel: + 420 702 137 233 | Magyarország CSL Behring Kft. Tel.: +36 1 213 4290 |
Danmark CSL Behring AB Tlf: +46 8 544 966 70 | Malta AM Mangion Ltd. Tel: +356 2397 6333 |
Deutschland CSL Behring GmbH Tel: +49 6190 75 84810 | Nederland CSL Behring BV Tel: + 31 85 111 96 00 |
Eesti CentralPharma Communications OÜ Tel: +3726015540 | Norge CSL Behring AB Tlf: +46 8 544 966 70 |
Ελλáδα CSL Behring ΕΠΕ Τηλ: +30 210 7255 660 | Österreich CSL Behring GmbH Tel: +43 1 80101 1040 |
España CSL Behring S.A. Tel: +34 933 67 1870 | Polska CSL Behring Sp.z o.o. Tel: +48 22 213 22 65 |
France CSL Behring S.A. Tél: + 33 –(0)-1 53 58 54 00 | Portugal CSL Behring Lda Tel: +351 21 782 62 30 |
Hrvatska Marti Farm d.o.o. Tel: +385 1 5588297 | România Prisum Healthcare S.R.L. Tel: +40 21 322 0171 |
Ireland CSL Behring GmbH Tel: +49 6190 75 84700 Ísland CSL Behring AB Sími: +46 8 544 966 70 | Slovenija Emmes Biopharma Global s.r.o. podružnica v Sloveniji Tel:+ 386 41 42 0002 Slovenská republika CSL Behring Slovakia s.r.o. Tel: +421 911 653 862 |
Italia CSL Behring S.p.A. Tel: +39 02 34964 200 | Suomi/Finland CSL Behring AB Puh/Tel: +46 8 544 966 70 |
Κúπρος CSL Behring ΕΠΕ Τηλ: +30 210 7255 660 | Sverige CSL Behring AB Tel: +46 8 544 966 70 |
Latvija CentralPharma Communications SIA Tel: +371 6 7450497 | |

Fecha de la última revisión de este prospecto: {12/2024}.
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
----------------------------------------------------------------------------------------------------------
Esta información está destinada únicamente a profesionales del sector sanitario:
Monitorización del tratamiento
Durante el transcurso del tratamiento, se recomienda controlar adecuadamente los niveles de factor VIII para determinar la dosis que se debe administrar y la frecuencia de las inyecciones. Las respuestas de los pacientes al factor VIII pueden variar, lo cual demuestra que posee distintas semividas y recuperaciones. Puede que la dosis basada en el peso corporal se deba ajustar en pacientes con peso insuficiente o sobrepeso. En el caso especial de las intervenciones de cirugía mayor, es indispensable controlar con precisión la terapia de sustitución mediante análisis de la coagulación (actividad del factor VIII plasmático).
Al utilizar un ensayo de coagulación de una etapa basado en el tiempo de tromboplastina parcial activada (TTPa) in vitropara determinar la actividad del factor VIII en las muestras de sangre de los pacientes, los resultados de la actividad del factor VIII plasmático pueden verse significativamente afectados tanto por el tipo de reactivo de TTPa como por el estándar de referencia utilizado en el ensayo. También se pueden producir discrepancias significativas entre los resultados obtenidos en el ensayo de coagulación de una etapa basado en el TTPa y los obtenidos en el ensayo cromogénico según la Farmacopea Europea. Esto resulta especialmente importante cuando se cambia el laboratorio o los reactivos que se utilizan en el ensayo.
La actividad del factor VIII plasmático en los pacientes que reciben AFSTYLA con un análisis cromogénico o un ensayo de coagulación de una etapa se debe controlar para orientar la dosis administrada y la frecuencia de las inyecciones repetidas.. El resultado del análisis cromogénico refleja con más precisión el potencial hemostático clínico de AFSTYLA, por lo que es el método preferido. El resultado del ensayo de coagulación de una etapa subestima el nivel de actividad del factor VIII en comparación con el resultado del ensayo cromogénico en aproximadamente un 45%. Si se utiliza un ensayo de coagulación de una etapa, se multiplica el resultado por un factor de conversión de 2 para determinar el nivel de actividad del factor VIII del paciente.
Posología
La dosis y la duración de la terapia de sustitución dependen de la gravedad de la deficiencia de factor VIII, de la localización y extensión de la hemorragia y del estado clínico del paciente.
El número de unidades de factor VIII administradas se expresa en Unidades Internacionales (UI), que se corresponden con el actual estándar concentrado de la OMS para medicamentos con factor VIII. La actividad del factor VIII en plasma se expresa como un porcentaje (en relación con el plasma humano normal) o, preferiblemente, en Unidades Internacionales (en relación con un estándar internacional para el factor VIII en plasma).
Una Unidad Internacional (UI) de actividad del factor VIII equivale a la cantidad de factor VIII presente en un ml de plasma humano normal.
La asignación de potencia se determina mediante un ensayo de sustratos cromogénicos.
Los niveles plasmáticos de factor VIII se pueden monitorizar mediante un ensayo de sustratos cromogénicos o un ensayo de coagulación de una etapa.
Tratamiento a demanda
El cálculo de la dosis necesaria de factor VIII se basa en el hallazgo empírico de que 1 Unidad Internacional (UI) de factor VIII por kg de peso corporal aumenta la actividad plasmática del factor VIII en 2 UI/dl.
La dosis requerida se determina usando la fórmula siguiente: Dosis (UI) = peso corporal (kg) x aumento deseado del factor VIII (UI/dl o % del nivel normal) x 0,5 (UI/kg por UI/dl)
La dosis y la frecuencia de administración se establecerán siempre en función de la eficacia clínica observada en cada caso.
En el caso de los acontecimientos hemorrágicos siguientes, la actividad del factor VIII no debe ser inferior al nivel de actividad plasmática establecido (en % del nivel normal o UI/dl) durante el período correspondiente. La tabla siguiente puede usarse como guía posológica en episodios hemorrágicos y cirugía:
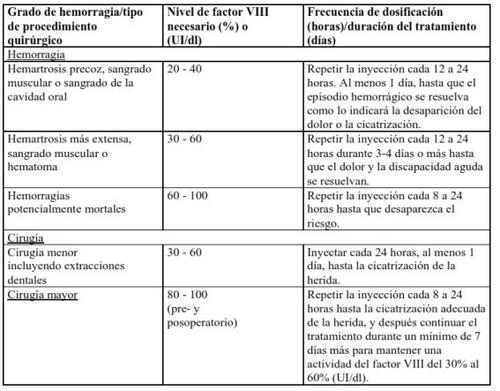
Tratamiento profiláctico
La pauta de tratamiento inicial recomendada es de 20 a 50 UI/kg de AFSTYLA administradas 2 o 3 veces a la semana. La pauta se puede ajustar en función de la respuesta del paciente.
Población pediátrica
La pauta de tratamiento inicial recomendada en niños (de 0 a < 12 años de edad) es de 30 a 50 UI por kg de AFSTYLA administradas 2 o 3 veces a la semana. Puede que en los niños < 12 años se requieran dosis más frecuentes o más altas debido al mayor aclaramiento que se presenta en este grupo de edad.
En los adolescentes con 12 años o más de edad, las dosis recomendadas son las mismas que para los adultos.
Población de edad avanzada
En los estudios clínicos de AFSTYLA no se incluyeron sujetos mayores de 65 años de edad
- País de registro
- Principio activo
- Requiere recetaSí
- Fabricante
- Esta información es de carácter general y no sustituye la consulta con un profesional sanitario.
- Alternativas a AFSTYLA 1.500 UI POLVO Y DISOLVENTE PARA SOLUCION INYECTABLEForma farmacéutica: INYECTABLE, 1.000 UIPrincipio activo: factor de coagulación VIIIFabricante: Takeda Manufacturing Austria AgRequiere recetaForma farmacéutica: INYECTABLE, 1.500 UIPrincipio activo: factor de coagulación VIIIFabricante: Takeda Manufacturing Austria AgRequiere recetaForma farmacéutica: INYECTABLE, 1000 UI - tras reconstitución en 2 ml de agua para inyectables la dosis es de 500 UI/mlPrincipio activo: factor de coagulación VIIIFabricante: Takeda Manufacturing Austria AgRequiere receta
Médicos online para AFSTYLA 1.500 UI POLVO Y DISOLVENTE PARA SOLUCION INYECTABLE
Comenta la dosis, los posibles efectos secundarios, interacciones, contraindicaciones o la revisión de receta de AFSTYLA 1.500 UI POLVO Y DISOLVENTE PARA SOLUCION INYECTABLE, sujeto a valoración médica y a la normativa local.









Preguntas frecuentes
