


Smofkabiven extra Nitrogen Ef
Consulta con un médico sobre la receta médica de Smofkabiven extra Nitrogen Ef



Cómo usar Smofkabiven extra Nitrogen Ef
Hoja de instrucciones del paquete: información para el usuario
SmofKabiven extra Nitrogen EF, emulsión para infusión
Es importante leer detenidamente el contenido de esta hoja de instrucciones antes de usar el medicamento, ya que contiene
información importante para el paciente.
- Debe conservar esta hoja de instrucciones para poder volver a leerla si es necesario.
- Si tiene alguna duda, debe consultar a su médico, farmacéutico o enfermera.
- Si el paciente experimenta algún efecto adverso, incluidos los efectos adversos no mencionados en esta hoja de instrucciones, debe informar a su médico, farmacéutico o enfermera. Véase el punto 4.
Índice de la hoja de instrucciones
- 1. Qué es SmofKabiven extra Nitrogen EF y para qué se utiliza
- 2. Información importante antes de usar SmofKabiven extra Nitrogen EF
- 3. Cómo usar SmofKabiven extra Nitrogen EF
- 4. Posibles efectos adversos
- 5. Cómo conservar SmofKabiven extra Nitrogen EF
- 6. Contenido del paquete y otra información
1. Qué es SmofKabiven extra Nitrogen EF y para qué se utiliza
SmofKabiven extra Nitrogen EF es una emulsión para infusión, administrada al paciente a través de un goteo (infusión intravenosa). El paquete del medicamento es una bolsa de plástico que contiene aminoácidos (componentes necesarios para la formación de proteínas), glucosa (carbohidratos) y grasas (lípidos). El medicamento puede ser utilizado en pacientes adultos y niños a partir de 2 años de edad.
El personal médico especializado administra SmofKabiven extra Nitrogen EF cuando otros métodos de nutrición son insuficientes o imposibles.
2. Información importante antes de usar SmofKabiven extra Nitrogen EF
No use SmofKabiven extra Nitrogen EF si el paciente tiene:
- alergia a los principios activos o a alguno de los demás componentes de este medicamento (enumerados en el punto 6);
- alergia al proteína de pescado o huevo;
- alergia a los frutos secos o soja (SmofKabiven extra Nitrogen EF contiene aceite de soja);
- concentración demasiado alta de grasas en la sangre (hiperlipidemia);
- trastornos graves de la función hepática;
- problemas de coagulación de la sangre (trastornos de la coagulación);
- trastorno del metabolismo de los aminoácidos;
- enfermedad renal grave, sin posibilidad de diálisis;
- shock agudo;
- concentración no controlada de glucosa en la sangre (hiperglucemia);
- líquido en los pulmones (edema pulmonar agudo);
- demasiado líquido en el cuerpo (sobrehidratación);
- insuficiencia cardíaca no tratada;
- trastorno de la coagulación de la sangre (síndrome hemofagocítico);
- estado general inestable, por ejemplo, trauma grave, diabetes no controlada, infarto de miocardio, accidente cerebrovascular, trombosis, acidosis metabólica (trastorno caracterizado por una cantidad excesiva de sustancias ácidas en la sangre), infección grave (sepsis grave), coma, déficit de líquido (deshidratación hipotónica).
No se debe usar SmofKabiven extra Nitrogen EF en recién nacidos y niños menores de 2 años.
Advertencias y precauciones
Antes de iniciar el tratamiento con SmofKabiven extra Nitrogen EF, debe discutir con su médico si el paciente tiene:
- enfermedad renal;
- diabetes;
- pancreatitis;
- enfermedad hepática;
- hipotiroidismo (trastornos de la tiroides);
- sepsis (infección grave).
Si durante la infusión se produce fiebre, erupción, hinchazón, dificultad para respirar, escalofríos, sudoración, náuseas o vómitos, debe informar inmediatamente a su médico, ya que estos síntomas pueden ser causados por una reacción alérgica o por una dosis demasiado alta del medicamento.
Su médico puede recomendar análisis de sangre regulares para determinar las pruebas de función hepática y otros valores.
Niños y adolescentes
SmofKabiven extra Nitrogen EF no está indicado para recién nacidos o niños menores de 2 años. SmofKabiven extra Nitrogen EF puede ser administrado a niños de 2 a 16/18 años.
SmofKabiven extra Nitrogen EF y otros medicamentos
Debe informar a su médico sobre todos los medicamentos que el paciente esté tomando actualmente o haya tomado recientemente, así como sobre los medicamentos que el paciente planea tomar, incluidos los que se venden sin receta.
Embarazo y lactancia
No hay datos sobre el uso de SmofKabiven extra Nitrogen EF durante el embarazo o la lactancia. SmofKabiven extra Nitrogen EF se administra a mujeres embarazadas o en período de lactancia solo si el médico lo considera necesario. SmofKabiven extra Nitrogen EF puede ser administrado durante el embarazo y la lactancia bajo prescripción médica.
Conducción de vehículos y uso de maquinaria
No aplica, ya que este medicamento se administra en el hospital.
3. Cómo usar SmofKabiven extra Nitrogen EF
Este medicamento debe ser utilizado siempre según las indicaciones de su médico. En caso de duda, debe consultar a su médico.
Su médico determinará la dosis individual según el peso y el estado clínico del paciente. SmofKabiven extra Nitrogen EF se administra solo por personal médico especializado.
Uso de una dosis mayor que la recomendada de SmofKabiven extra Nitrogen EF
Es poco probable que el paciente reciba una dosis demasiado alta de SmofKabiven extra Nitrogen EF, ya que el medicamento se administra por personal médico especializado.
4. Posibles efectos adversos
Como cualquier medicamento, este medicamento puede causar efectos adversos, aunque no todos los pacientes los experimentarán.
Efectos adversos frecuentes(pueden ocurrir en hasta 1 de cada 10 pacientes):
ligera fiebre.
Efectos adversos poco frecuentes(pueden ocurrir en hasta 1 de cada 100 pacientes): alta concentración de enzimas hepáticos en la sangre, pérdida de apetito, náuseas, vómitos, escalofríos, mareos y dolor de cabeza.
Efectos adversos raros(pueden ocurrir en hasta 1 de cada 1000 pacientes): baja o alta presión arterial, dificultad para respirar, ritmo cardíaco acelerado (taquicardia).
Reacciones de hipersensibilidad (que pueden causar síntomas como hinchazón, fiebre, caída de la presión arterial, erupción cutánea, ampollas, enrojecimiento, dolor de cabeza). Sensación de calor y frío. Palidez. Leve cianosis de los labios y la piel (relacionada con la hipoxia). Dolor en el cuello, la espalda, los huesos, el pecho y la región lumbar.
Notificación de efectos adversos
Si se producen algún efecto adverso, incluidos los efectos adversos no mencionados en esta hoja de instrucciones, debe informar a su médico, farmacéutico o enfermera. Los efectos adversos pueden notificarse directamente al Departamento de Vigilancia de Medicamentos del Ministerio de Sanidad, Consumo y Bienestar Social
Calles de Alcalá, 56
28071 Madrid
Teléfono: +34 91 596 24 99, fax: +34 91 596 24 90
Sitio web: https://www.aemps.gob.es/
Los efectos adversos también pueden notificarse al titular de la autorización de comercialización.
La notificación de efectos adversos permite recopilar más información sobre la seguridad del medicamento.
5. Cómo conservar SmofKabiven extra Nitrogen EF
El medicamento debe conservarse en un lugar donde no pueda ser visto o alcanzado por los niños.
No conservar a una temperatura superior a 25°C. No congelar. Conservar en la bolsa exterior.
No usar este medicamento después de la fecha de caducidad indicada en la bolsa y la caja de cartón.
La fecha de caducidad es el último día del mes indicado.
6. Contenido del paquete y otra información
Qué contiene SmofKabiven extra Nitrogen EF?
Los principios activos del medicamento son:
g/1000 ml
alanina
9,2
arginina
7,9
glicina
7,2
histidina
2,0
isoleucina
3,3
leucina
4,8
lisina (como acetato)
4,3
metionina
2,8
fenilalanina
3,3
prolina
7,3
serina
4,3
taurina
0,65
treonina
2,9
triptófano
1,3
tirosina
0,26
valina
4,1
glucosa (en forma de monohidrato)
85
aceite de soja refinado
8,7
triglicéridos de ácidos grasos saturados de cadena media
8,7
aceite de oliva refinado
7,2
aceite de pescado rico en ácidos omega-3
4,3
Los demás componentes (excipientes) son: glicerol, fosfolipidos de huevo purificados, alfa-tocoferol, hidróxido de sodio (para ajustar el pH), oleato de sodio, ácido acético glacial (para ajustar el pH), ácido clorhídrico (para ajustar el pH) y agua para inyección.
Cómo se presenta SmofKabiven extra Nitrogen EF y qué contiene el paquete?
Las soluciones de glucosa y aminoácidos son transparentes, incoloras a ligeramente amarillas, sin partículas sólidas. La emulsión de grasas es blanca y homogénea.
Tamaños del paquete:
1 × 506 ml, 6 × 506 ml
1 × 1012 ml, 4 × 1012 ml
1 × 1518 ml, 4 × 1518 ml
1 × 2025 ml, 4 × 2025 ml
1 × 2531 ml, 3 × 2531 ml
No todos los tamaños del paquete deben estar disponibles en el mercado.
Título de la autorización de comercialización y fabricante
Fresenius Kabi AB
Rapsgatan 7
751 74 Uppsala
Suecia
Para obtener más información, debe consultar a un representante del titular de la autorización de comercialización:
Fresenius Kabi España, S.A.
Avda. de la Industria, 32
28108 Alcobendas (Madrid)
Teléfono: +34 91 657 43 00
Fecha de la última revisión de la hoja de instrucciones:09.06.2023
---------------------------------------------------------------------------------------------------------------------------
Información destinada exclusivamente a profesionales de la salud:
Advertencias y precauciones para la administración
Para evitar riesgos asociados con la infusión a una velocidad mayor que la recomendada, se recomienda realizarla de manera continua y controlada, siempre que sea posible, utilizando una bomba de volumen.
Como el uso de una vena central para la infusión conlleva un mayor riesgo de infección, durante la colocación y el manejo del catéter se recomienda seguir estrictamente las normas de asepsia para evitar cualquier infección.
Se recomienda controlar la concentración de glucosa y electrolitos en suero, la osmolalidad y el balance de líquidos y el equilibrio ácido-base, así como realizar pruebas enzimáticas hepáticas.
En caso de aparición de cualquier signo o síntoma de reacción anafiláctica (como fiebre, escalofríos, erupción o dificultad para respirar), debe interrumpir inmediatamente la infusión.
No debe administrar SmofKabiven extra Nitrogen EF junto con sangre en el mismo conjunto de infusión, debido al riesgo de pseudoaglutinación.
Vía de administración
Administración intravenosa, infusión en vena central.
Para asegurar una nutrición parenteral completa, debe agregar a SmofKabiven extra Nitrogen EF oligoelementos, electrolitos y vitaminas, según las necesidades del paciente.
Dosis
Pacientes adultos
Dosis recomendada
El rango de dosis es de 13 a 31 ml de SmofKabiven extra Nitrogen EF/kg de peso corporal/día, lo que proporciona de 0,14 a 0,32 g de nitrógeno/kg de peso corporal/día (de 0,85 a 2,0 g de aminoácidos/kg de peso corporal/día) y de 12 a 28 kcal/kg de peso corporal/día de energía total (de 8 a 19 kcal/kg de peso corporal/día de energía no proteica).
Velocidad de infusión
La velocidad máxima de infusión de glucosa es generalmente de 0,25 g/kg de peso corporal/hora, de aminoácidos 0,1 g/kg de peso corporal/hora, y de grasas 0,15 g/kg de peso corporal/hora.
La velocidad de infusión no debe ser mayor que 1,5 ml/kg de peso corporal/hora (lo que equivale a 0,13 g de glucosa, 0,10 g de aminoácidos y 0,04 g de grasas/kg de peso corporal/hora). El tiempo de infusión recomendado es de 14 a 24 horas.
Nutrición parenteral durante la diálisis (IDPN, por sus siglas en inglés)
En pacientes adultos clínicamente estables sometidos a terapia de reemplazo renal crónica, la velocidad máxima de infusión en la nutrición parenteral durante la diálisis (IDPN) es de 3,0 ml/kg de peso corporal/hora (lo que equivale a 0,20 g/kg de peso corporal/hora de aminoácidos, 0,25 g/kg de peso corporal/hora de glucosa y 0,09 g/kg de peso corporal/hora de grasas). El volumen de infusión en la IDPN debe depender de la diferencia entre la ingesta oral de alimentos y la ingesta recomendada de nutrientes, las pérdidas inevitables de nutrientes causadas por el tratamiento de reemplazo renal, así como de la tolerancia metabólica individual del paciente. El tiempo de infusión en la IDPN suele ser de 3 a 5 horas, dependiendo de las necesidades del paciente y del tiempo programado para la sesión de tratamiento de reemplazo renal. La dosis diaria máxima recomendada sigue siendo la misma.
Dosis diaria máxima
La dosis diaria máxima depende del estado clínico del paciente y puede cambiar incluso de un día a otro. La dosis diaria máxima recomendada es de 31 ml/kg de peso corporal/día.
Niños y adolescentes
Niños de 2 a 11 años
Dosis recomendada
La dosis de hasta 31 ml/kg de peso corporal/día debe ajustarse regularmente a las necesidades del paciente en edad pediátrica, que varían mucho más que en los pacientes adultos.
Velocidad de infusión
La velocidad máxima de infusión recomendada es de 1,8 ml/kg de peso corporal/hora (lo que equivale a 0,12 g de aminoácidos/kg de peso corporal/hora, 0,15 g de glucosa/kg de peso corporal/hora y 0,05 g de grasas/kg de peso corporal/hora).
Excepto en situaciones especiales que requieren un seguimiento cuidadoso, cuando se utiliza la velocidad máxima de infusión recomendada, el tiempo de infusión no debe exceder las 17 horas.
El tiempo de infusión recomendado es de 12 a 24 horas.
Dosis diaria máxima
La dosis diaria máxima es variable según el estado clínico del paciente y puede cambiar incluso de un día a otro. La dosis diaria máxima recomendada es de 31 ml/kg de peso corporal/día.
Adolescentes de 12 a 16/18 años
En adolescentes, SmofKabiven extra Nitrogen EF puede ser dosificado como en pacientes adultos.
Precauciones especiales para la eliminación y preparación del medicamento para su uso
No use si el paquete está dañado.
Use solo si las soluciones de aminoácidos y glucosa son transparentes, incoloras a ligeramente amarillas, y la emulsión de grasas es blanca y homogénea .El contenido de las tres cámaras separadas debe mezclarse antes de su uso, así como antes de agregar otros componentes a través del puerto designado para este propósito.
Después de retirar las cubiertas de protección, debe girar el bolso varias veces para mezclar completamente todos los componentes del medicamento y obtener una mezcla homogénea en la que no se puedan ver signos de separación de fases.
Solo para uso único. Todos los restos no utilizados del medicamento que queden después de la infusión deben ser destruidos.
Compatibilidad
Los datos sobre la compatibilidad están disponibles para los medicamentos Dipeptiven, Addamel N/Supliven, Glycophos, Addiphos, Vitalipid N Adult/Infant y Soluvit N en cantidades y concentraciones específicas de electrolitos.
Al agregar electrolitos, debe tener en cuenta las cantidades ya presentes en el bolso para satisfacer las necesidades clínicas del paciente. Los datos disponibles confirman la posibilidad de agregar los medicamentos mencionados anteriormente al bolso activado de acuerdo con la siguiente tabla:
Alcance de la compatibilidad: estable durante 7 días, es decir, 6 días almacenado a una temperatura de 2-8 °C, y luego 24 horas a una temperatura de 20-25 °C.
| Unidad | Cantidad máxima total | |||||
| Tamaño del bolso de SmofKabiven extra Nitrogen EF | ml | 506 | 1012 | 1518 | 2025 | 2531 |
| Agregar | Volumen | |||||
| Dipeptiven | ml |
|
|
|
|
|
| Supliven/Addamel N | ml |
|
|
|
|
|
| Soluvit N | ampolla(s) |
|
|
|
|
|
| Vitalipid N Adult/Infant | ml |
|
|
|
|
|
| Límites de electrolitos1 | Concentración | |||||
| Sodio | mmol/l | ≤ 150 | ≤ 150 | ≤ 150 | ≤ 150 | ≤ 150 |
| Potasio | mmol/l | ≤ 150 | ≤ 150 | ≤ 150 | ≤ 150 | ≤ 150 |
| Calcio | mmol/l | ≤ 5 | ≤ 5 | ≤ 5 | ≤ 5 | ≤ 5 |
| Magnesio | mmol/l | ≤ 5 | ≤ 5 | ≤ 5 | ≤ 5 | ≤ 5 |
| Fosfato inorgánico (Addiphos) o fosfato orgánico (Glycophos) | mmol/l | ≤ 15 ≤ 30 | ≤ 15 ≤ 30 | ≤ 15 ≤ 30 | ≤ 15 ≤ 30 | ≤ 15 ≤ 30 |
| Cinc | mmol/l | ≤ 0,2 | ≤ 0,2 | ≤ 0,2 | ≤ 0,2 | ≤ 0,2 |
| Selenio | μmol/l | ≤ 2 | ≤ 2 | ≤ 2 | ≤ 2 | ≤ 2 |
Nota: esta tabla tiene como objetivo mostrar la compatibilidad. No constituye una guía para la dosificación.
Antes de prescribir los medicamentos mencionados, debe consultar las hojas de instrucciones aprobadas.
La información sobre la compatibilidad con otros aditivos y los tiempos de almacenamiento de diferentes mezclas estará disponible a petición.
Todos los aditivos deben mezclarse con el medicamento en condiciones asépticas.
Periodo de validez después de mezclar el contenido de las cámaras del bolso
Se ha demostrado la estabilidad física y química del contenido mezclado del bolso de tres cámaras durante 48 horas a una temperatura de 20-25 °C. Desde el punto de vista microbiológico, el medicamento debe usarse inmediatamente.
En caso contrario, el usuario es responsable del período de almacenamiento durante el uso y de las condiciones de almacenamiento antes de la administración. Este período no debe exceder normalmente las 24 horas a una temperatura de 2-8 °C, a menos que la mezcla se haya realizado en condiciones asépticas controladas y validadas.
Periodo de validez después de mezclar con sustancias adicionales
Se ha demostrado la estabilidad físico-química del contenido mezclado del bolso de tres cámaras con sustancias adicionales durante un período de hasta 7 días, es decir, 6 días a una temperatura de 2-8 °C, y luego 24 horas a una temperatura de 20-25 °C, incluido el tiempo de infusión. Desde el punto de vista microbiológico, el medicamento debe usarse inmediatamente después de agregar otros componentes. En caso contrario, el usuario es responsable del período de almacenamiento durante el uso y de las condiciones de almacenamiento antes de la administración. Este período no debe exceder normalmente las 24 horas a una temperatura de 2-8 °C, a menos que la mezcla se haya realizado en condiciones asépticas controladas y validadas.
SmofKabiven extra Nitrogen EF Instrucciones para la preparación del bolso para su uso
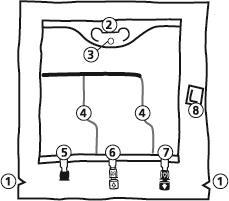
- 1. Corte en el bolso exterior
- 2. Asa del bolso
- 3. Orificio de suspensión del bolso
- 4. Soldaduras que separan las cámaras del bolso
- 5. Puerto ciego (solo se utiliza en la producción)
- 6. Puerto para la administración de sustancias adicionales
- 7. Puerto de infusión
- 8. Absorbente de oxígeno
| |
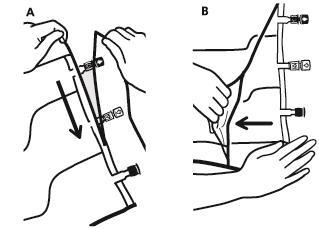 | |
- Para retirar el bolso exterior, debe colocarlo horizontalmente y, comenzando por el corte que se encuentra cerca de los puertos, rasgar a lo largo del borde superior (A).
- Luego, rasgar el bolso exterior a lo largo del borde largo, quitarlo y desecharlo junto con el absorbente de oxígeno (B).
2. Mezcla
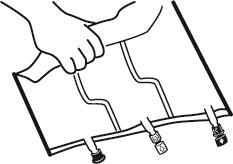
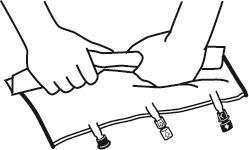
- Colocar el bolso en una superficie plana.
- Comenzando desde el lado de la asa, enrollar firmemente el bolso hacia los puertos, primero con la mano derecha y luego aplicando una presión constante con la mano izquierda, hasta que se abran las soldaduras verticales. Se abren bajo la presión del líquido. Las soldaduras también se pueden abrir antes de retirar el bolso exterior. Nota:el líquido se mezcla fácilmente, aunque la soldadura horizontal permanece intacta.
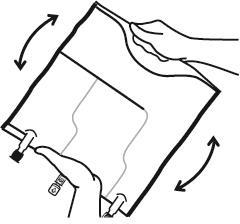
- Mezclar el contenido de las tres cámaras girando tres veces el bolso, lo que debe asegurar una mezcla completa de los componentes.
| |
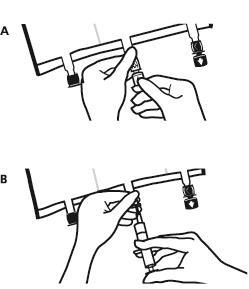 | |
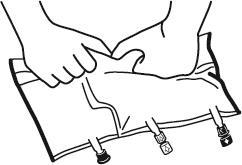
- Volver a colocar el bolso en una superficie plana y llana. Inmediatamente antes de agregar sustancias adicionales, quitar el tapón de un solo uso que cubre el puerto blanco para la administración de sustancias adicionales (A).
Nota:la membrana del puerto para la administración de sustancias adicionales es estéril.
- Sostener la base del puerto para la administración de sustancias adicionales. Introducir la aguja, inyectar las sustancias adicionales (con una compatibilidad conocida) a través del centro del lugar de inyección (B).
- Mezclar cuidadosamente el contenido del bolso después de agregar cada componente girando tres veces el bolso después de cada adición. Utilizar jeringas con agujas de 18 a 23 G y una longitud máxima de 40 mm.
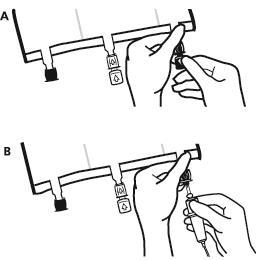
- Inmediatamente antes de conectar el conjunto de infusión, quitar el tapón de un solo uso que cubre el puerto de infusión azul (A). Nota:la membrana del puerto de infusión es estéril.
- Utilizar conjuntos de infusión sin aireador o cerrar el aireador.
- Sostener la base del puerto de infusión.
- Introducir la aguja del conjunto de infusión en el puerto de infusión. Para asegurar una buena fijación de la aguja, debe introducir toda su longitud. Nota:la superficie interna del puerto de infusión es estéril.
4. Suspensión del bolso
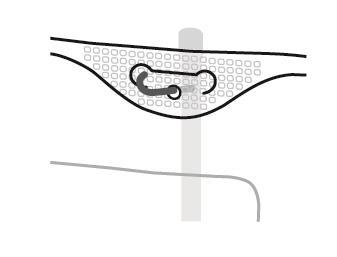
- Suspender el bolso utilizando el orificio que se encuentra debajo de la asa.
- País de registro
- Principio activo
- Requiere recetaSí
- Fabricante
- ImportadorFresenius Kabi AB
- Esta información ha sido traducida con IA y es solo orientativa. No constituye asesoramiento médico. Consulta siempre con un médico antes de tomar cualquier medicamento.
- Alternativas a Smofkabiven extra Nitrogen EfForma farmacéutica: Solución, -Principio activo: combinationsNo requiere recetaForma farmacéutica: Solución, -Principio activo: combinationsNo requiere recetaForma farmacéutica: Solución, -Principio activo: combinationsNo requiere receta
Alternativas a Smofkabiven extra Nitrogen Ef en otros países
Las mejores alternativas con el mismo principio activo y efecto terapéutico.
Alternativa a Smofkabiven extra Nitrogen Ef en Hiszpania
Alternativa a Smofkabiven extra Nitrogen Ef en Ukraina
Médicos online para Smofkabiven extra Nitrogen Ef
Consulta sobre dosis, efectos secundarios, interacciones, contraindicaciones y renovación de la receta de Smofkabiven extra Nitrogen Ef – sujeta a valoración médica y normativa local.









Mantente informado y ahorra en salud
Recibe consejos de salud, novedades de la plataforma y promociones exclusivas para suscriptores.

