

Ig Vena
Consulta con un médico sobre la receta médica de Ig Vena



Cómo usar Ig Vena
Hoja de instrucciones incluida en el paquete: información para el usuario
Ig VENA, 50 g/l, solución para infusión
Inmunoglobulina humana normal para administración intravenosa (IVIg)
Es importante que lea este prospecto detenidamente antes de tomar este medicamento, ya que contiene información importante para usted.
- Debe conservar este prospecto para poder volver a leerlo más adelante.
- Si tiene alguna duda, consulte a su médico o enfermera.
- Si el paciente presenta algún síntoma adverso, incluidos los síntomas adversos no mencionados en este prospecto, debe informar a su médico o enfermera. Véase el apartado 4.
Índice
- 1. Qué es Ig VENA y para qué se utiliza
- 2. Información importante antes de tomar Ig VENA
- 3. Cómo tomar Ig VENA
- 4. Efectos adversos
- 5. Conservación de Ig VENA
- 6. Contenido del paquete y otra información
1. Qué es Ig VENA y para qué se utiliza
Ig VENA es una solución de inmunoglobulina humana normal para administración intravenosa.
Las inmunoglobulinas son anticuerpos humanos presentes en la sangre.
Ig VENA se utiliza en los siguientes tratamientos:
Tratamiento de adultos y niños y adolescentes (0-18 años) cuando el paciente no tiene suficientes
anticuerpos (tratamiento de sustitución) en los siguientes casos:
- 1. En pacientes con deficiencia congénita de producción de anticuerpos (en síndromes de deficiencia primaria de la inmunidad).
- 2. En pacientes con deficiencia adquirida de producción de anticuerpos (deficiencia secundaria de la inmunidad), que presentan infecciones graves o recurrentes causadas por diferentes condiciones clínicas (por ejemplo, enfermedades oncológicas o autoinmunes, o como resultado del tratamiento de estas enfermedades). El tratamiento con antibióticos en estos pacientes no ha sido efectivo y, o no han obtenido un aumento suficiente de los títulos de anticuerpos IgG después de la vacunación (vacuna antineumocócica polisacárida y vacuna que contiene antígeno polipéptido), o el nivel de IgG en suero es inferior a 4 g/l.
Tratamiento de adultos y niños y adolescentes (0-18 años) con ciertas enfermedades inflamatorias (inmunomodulación) en los siguientes casos:
- 1. En pacientes con un número insuficiente de plaquetas (púrpura trombocitopénica idiopática) y en pacientes con alto riesgo de sangrado o antes de una operación para obtener un número adecuado de plaquetas.
- 2. En pacientes con síndrome de Guillain-Barré. Es una enfermedad aguda que se caracteriza por inflamación de los nervios periféricos, lo que causa una debilidad muscular grave, principalmente en las piernas y extremidades superiores.
- 3. En pacientes con enfermedad de Kawasaki (en combinación con ácido acetilsalicílico). La enfermedad de Kawasaki es una enfermedad aguda que afecta principalmente a niños pequeños y se caracteriza por inflamación de los vasos sanguíneos de todo el cuerpo.
- 4. En pacientes con polineuropatía inflamatoria desmielinizante crónica (CIDP, por sus siglas en inglés). Esta enfermedad crónica es un trastorno raro de los nervios periféricos que se caracteriza por una debilidad muscular progresiva de las extremidades inferiores y, en menor medida, de las extremidades superiores.
- 5. En polineuropatía motora multifocal (MMN, por sus siglas en inglés). Es una enfermedad rara que afecta los nervios motores y se caracteriza por una debilidad muscular progresiva y asimétrica de las extremidades sin pérdida de sensibilidad.
2. Información importante antes de tomar Ig VENA
Cuándo no tomar Ig VENA
Si el paciente es alérgico (hipersensible) a la inmunoglobulina humana normal o a alguno de los demás componentes de este medicamento (enumerados en el apartado 6).
Cuando el paciente tiene anticuerpos contra la inmunoglobulina A (IgA) en la sangre, ya que la administración de un medicamento que contiene IgA puede provocar una reacción alérgica grave.
Advertencias y precauciones
Antes de comenzar a tomar Ig VENA, debe hablar con su médico o enfermera.
Los pacientes deben ser monitorizados y observados cuidadosamente durante la infusión debido al riesgo de efectos adversos.
Algunas reacciones adversas pueden ocurrir con más frecuencia:
- en caso de infusión demasiado rápida;
- en pacientes con signos de infección no tratada (por ejemplo, fiebre) o con una condición inflamatoria crónica;
- en pacientes que reciben inmunoglobulina humana normal por primera vez;
- en casos raros, cuando se cambia el medicamento de inmunoglobulina humana normal que se administraba previamente o cuando ha pasado mucho tiempo desde la última infusión.
En algunos casos, las inmunoglobulinas pueden aumentar el riesgo de infarto de miocardio, accidente cerebrovascular, trombosis pulmonar o empeorar la trombosis venosa profunda
Por este motivo, el médico debe tener especial cuidado en los siguientes casos:
- en pacientes obesos,
- en personas de edad avanzada,
- en pacientes con diabetes,
- en pacientes con hipertensión,
- en pacientes con volumen sanguíneo reducido (hipovolemia),
- en pacientes con enfermedades vasculares,
- en pacientes con riesgo de trombosis sanguínea (trastornos adquiridos o congénitos de la coagulación),
- en pacientes con antecedentes de trombosis,
- en pacientes con enfermedades que se caracterizan por un aumento de la viscosidad sanguínea,
- en pacientes inmovilizados durante mucho tiempo,
- en pacientes con enfermedades renales actuales o pasadas, o que toman medicamentos que pueden dañar los riñones (medicamentos nefrotóxicos), ya que se han registrado casos de insuficiencia renal aguda. En caso de daño renal, debe considerarse la interrupción de la administración de inmunoglobulina.
El paciente puede ser alérgico (hipersensible) a la inmunoglobulina (anticuerpos) sin saberlo
La hipersensibilidad puede ocurrir incluso en pacientes que han recibido previamente inmunoglobulina humana normal y la han tolerado bien. Puede ocurrir especialmente en casos de deficiencia de inmunoglobulina tipo IgA (en pacientes con anticuerpos anti-IgA). En estos casos raros, pueden ocurrir reacciones alérgicas (hipersensibilidad) como una caída repentina de la presión arterial o una reacción anafiláctica.
En caso de una reacción adversa, debe reducirse la velocidad de administración o interrumpirse la administración de inmunoglobulina. El tratamiento depende del tipo y la gravedad de la reacción adversa. En caso de anafilaxia, debe procederse según los estándares médicos actuales para el tratamiento de la anafilaxia. Debe informar a su médico si alguno de los casos anteriores se aplica a usted. El médico tomará las medidas de precaución adecuadas al administrar Ig VENA.
Prevención de infecciones virales
Los medicamentos producidos a partir de sangre o plasma humanos se someten a procedimientos específicos para evitar la transmisión de infecciones a los pacientes tratados. Estos procedimientos incluyen la selección de donantes de sangre y plasma para excluir a los donantes que puedan ser una fuente de infección; la prueba de plasma para detectar la presencia de agentes infecciosos/virus. Los fabricantes de medicamentos a partir de sangre o plasma humanos también utilizan procesos que inactivan o eliminan los virus. A pesar de estas medidas de prevención, no se puede excluir completamente la posibilidad de transmisión de agentes infecciosos por el medicamento preparado a partir de sangre o plasma humano. Esto se aplica también a los virus desconocidos o recientemente descubiertos y otros patógenos.
Se considera que las medidas de prevención adoptadas son efectivas contra los virus envueltos, como el virus de la inmunodeficiencia humana (VIH), el virus de la hepatitis B (VHB) y el virus de la hepatitis C (VHC), así como contra los virus no envueltos, como el virus de la hepatitis A (VHA).
Estas medidas pueden tener una eficacia limitada contra los virus no envueltos, como el parvovirus B19.
No se ha demostrado que las inmunoglobulinas sean la causa de la hepatitis A o la infección por parvovirus B19, ya que la presencia de anticuerpos puede desempeñar un papel importante en la protección contra las infecciones virales.
Se recomienda registrar el nombre y el número de lote de Ig VENA cada vez que se administra, para permitir la identificación del medicamento.
Niños y adolescentes
Después de la administración de Ig VENA a niños y adolescentes, se ha observado glucosuria transitoria y leve (presencia de glucosa en la orina), sin síntomas clínicos. Esto puede estar relacionado con la presencia de maltosa en el medicamento Ig VENA, que se hidroliza a glucosa en los túbulos renales. La glucosa se reabsorbe y se elimina en la orina en muy pequeña medida. La reabsorción de glucosa depende de la edad del paciente. El aumento transitorio de la concentración de maltosa en el suero puede exceder la capacidad de reabsorción renal de azúcar y afectar el resultado de la prueba de glucosa en la orina.
Ig VENA y otros medicamentos
Debe informar a su médico sobre todos los medicamentos que esté tomando actualmente o recientemente, así como sobre los medicamentos que planea tomar.
No se debe mezclar la inmunoglobulina humana normal para administración intravenosa con otros medicamentos o con otros medicamentos que contengan inmunoglobulinas para administración intravenosa (IVIg).
Efecto en las vacunas vivas atenuadas
La administración de inmunoglobulina puede reducir la eficacia de las vacunas que contienen virus vivos atenuados, como la vacuna contra el sarampión, la rubéola, la varicela o la varicela, durante al menos 6 semanas y hasta 3 meses. Después de la administración de este medicamento, debe esperar 3 meses antes de vacunarse con una vacuna que contenga virus vivos atenuados. En el caso del sarampión, esta reducción de la eficacia puede durar hasta un año. Por lo tanto, los pacientes que reciben la vacuna contra el sarampión deben tener un título de anticuerpos determinado.
Diuréticos de asa (grupo de medicamentos que aumentan la producción de orina)
Debe evitar el uso concomitante con diuréticos de asa.
Efecto en los resultados de las pruebas de sangre
Después de la inyección de inmunoglobulina, el aumento transitorio en la sangre del paciente de diferentes anticuerpos pasivamente transferidos puede causar resultados falsos positivos en las pruebas serológicas.
La transferencia pasiva de anticuerpos contra antígenos de los glóbulos rojos, como A, B, D (responsables del grupo sanguíneo), puede afectar los resultados de algunas pruebas serológicas para anticuerpos contra los glóbulos rojos, como la prueba de anticuerpos directa (DAT, prueba de Coombs).
Prueba de glucosa en sangre
Algunos tipos de pruebas de glucosa en sangre (por ejemplo, los que utilizan métodos basados en la deshidrogenasa de la glucosa - piroloquinolina quinona (GDH-PQQ) o la oxidoreductasa de la glucosa - reactivo) interpretan falsamente la maltosa (100 mg/ml) presente en el medicamento Ig VENA como glucosa. Esto puede causar una lectura falsamente elevada de la concentración de glucosa en sangre durante la infusión y durante aproximadamente 15 horas después de la infusión, lo que puede llevar a una administración inadeuada de insulina, lo que puede provocar una hipoglucemia peligrosa para la vida. Además, los casos de hipoglucemia real pueden pasar desapercibidos si el estado de hipoglucemia está enmascarado por lecturas falsamente elevadas de la concentración de glucosa. En este contexto, al administrar Ig VENA o otros medicamentos para administración parenteral que contengan maltosa, la medición de la concentración de glucosa en sangre debe realizarse mediante un método específico para la glucosa. Debe leer cuidadosamente la información proporcionada con las tiras reactivas para la prueba de glucosa en sangre, incluida la información sobre las tiras reactivas, para determinar si se pueden utilizar con medicamentos para administración parenteral que contengan maltosa. En caso de duda, debe ponerse en contacto con el fabricante del dispositivo para determinar si se pueden utilizar con medicamentos para administración parenteral que contengan maltosa.
Niños y adolescentes
Aunque no se han realizado estudios de interacción en niños y adolescentes, no se espera que haya diferencias entre la población adulta y los niños y adolescentes.
Embarazo, lactancia y fertilidad
- Si la paciente está embarazada o en período de lactancia, cree que puede estar embarazada o planea tener un hijo, debe consultar a su médico antes de tomar este medicamento. El médico decidirá si Ig VENA puede administrarse a la paciente embarazada.
- No se han realizado estudios clínicos con Ig VENA en mujeres embarazadas. Se ha demostrado que los productos de inmunoglobulina intravenosa atraviesan la placenta, con mayor intensidad durante el tercer trimestre. Sin embargo, la experiencia clínica a largo plazo sobre el uso de inmunoglobulinas sugiere que no se debe esperar ningún efecto nocivo en el embarazo, el feto o el recién nacido.
- Si la paciente está en período de lactancia y recibe Ig VENA, los anticuerpos de este medicamento pueden pasar a la leche materna. Esto puede contribuir a la protección del lactante contra ciertas infecciones.
- La experiencia clínica sobre el uso de inmunoglobulinas sugiere que no se debe esperar ningún efecto nocivo en la fertilidad.
Conducción de vehículos y uso de máquinas
Algunos efectos adversos asociados con Ig VENA pueden empeorar la capacidad de conducir vehículos y utilizar máquinas. Los pacientes que hayan experimentado efectos adversos durante el tratamiento deben esperar a que desaparezcan antes de conducir un vehículo o utilizar máquinas.
Ig VENA contiene maltosa y sodio
El medicamento contiene 100 mg de maltosa en 1 ml.
Este medicamento contiene aproximadamente 69 mg de sodio en 1 litro. Debe tenerse en cuenta en pacientes con dieta baja en sodio.
3. Cómo tomar Ig VENA
Ig VENA solo puede ser administrado por un médico o personal médico capacitado en un entorno hospitalario o ambulatorio.
La dosis y el esquema de dosificación dependen de las indicaciones; el médico determinará la dosificación adecuada para cada paciente.
Ig VENA debe administrarse inicialmente de forma lenta. Si el medicamento es bien tolerado, la velocidad de infusión puede aumentarse gradualmente.
Uso en niños y adolescentes
La dosificación en niños y adolescentes (0-18 años) no difiere de la dosificación en adultos, ya que la dosificación en cada indicación se determina en función del peso corporal y la condición clínica del paciente.
Administración de una dosis mayor de la recomendada de Ig VENA
La sobredosis puede provocar sobrecarga circulatoria y viscosidad sanguínea excesiva, especialmente en pacientes con factores de riesgo, pacientes de edad avanzada o con insuficiencia cardíaca o renal.
En caso de dudas adicionales sobre la administración de este medicamento, debe consultar a su médico o enfermera.
4. Efectos adversos
Como cualquier medicamento, Ig VENA puede causar efectos adversos, aunque no todos los pacientes los experimentan.
Los siguientes efectos adversos pueden ocurrir después de la administración de un medicamento que contiene inmunoglobulina:
- escalofríos, dolor de cabeza, mareo, fiebre, vómitos, náuseas, reacciones alérgicas, dolor articular, hipotensión y dolor lumbar moderado pueden ocurrir ocasionalmente;
- casos aislados de reducción transitoria del recuento de glóbulos rojos (anemia hemolítica reversible/hemólisis);
- una caída repentina de la presión arterial puede ocurrir raramente, y en casos aislados, puede ocurrir un shock anafiláctico, incluso en pacientes que no han presentado hipersensibilidad en administraciones previas;
- se han observado reacciones cutáneas transitorias en casos raros;
- en casos muy raros, se han registrado complicaciones tromboembólicas (formación de coágulos), que pueden provocar infarto de miocardio, accidente cerebrovascular, trombosis pulmonar o trombosis venosa profunda;
- se han observado casos de meningitis aséptica (inflamación de las meninges sin infección);
- se han registrado aumentos del nivel de creatinina en suero y/o insuficiencia renal aguda;
- se han registrado casos de lesión pulmonar aguda relacionada con la transfusión (TRALI, por sus siglas en inglés).
En los estudios clínicos y después de la comercialización de Ig VENA, se han observado los siguientes efectos adversos, enumerados por frecuencia decreciente.
Frecuentes (pueden afectar hasta 1 de cada 10 personas)
- Dolor de espalda
- Náuseas
- Debilidad, fatiga, fiebre
- Dolor muscular
- Dolor de cabeza, somnolencia
Frecuencia desconocida (no puede estimarse a partir de los datos disponibles)
- Meningitis aséptica
- Hemólisis que causa anemia
- Reacciones alérgicas y shock anafiláctico potencialmente mortal
- Estado de confusión
- Accidente cerebrovascular, mareo, movimientos involuntarios, entumecimiento o hormigueo de la piel o las extremidades
- Infarto de miocardio, enfriamiento de la piel, taquicardia, bradicardia, ritmo cardíaco irregular
- Trombosis venosa profunda y trombosis arterial, hipotensión, hipertensión, palidez
- Émbolo pulmonar, edema pulmonar, dificultad para respirar con respiración sibilante o tos
- Vómitos, diarrea, dolor abdominal
- Erupciones cutáneas, urticaria, enrojecimiento y inflamación de la piel, erupción cutánea, picazón, eccema, sudoración excesiva
- Dolor articular y muscular, dolor de espalda, dolor de cuello, rigidez muscular
- Insuficiencia renal aguda
- Fiebre, dolor o molestia en el pecho, enrojecimiento de la cara, malestar general
- Aumento de la creatinina en sangre
Efectos adversos adicionales en niños y adolescentes
Debe esperarse que la frecuencia, tipo y gravedad de los efectos adversos en niños y adolescentes sean similares a los de los adultos.
Después de la administración de Ig VENA, en niños y adolescentes se ha observado glucosuria transitoria y leve (presencia de glucosa en la orina), sin síntomas clínicos.
Para obtener información sobre la seguridad viral, véase el apartado 2 "Información importante antes de tomar Ig VENA".
Notificación de efectos adversos
Si se producen efectos adversos, incluidos los efectos adversos no mencionados en este prospecto, debe informar a su médico o enfermera. Los efectos adversos pueden notificarse al Departamento de Vigilancia de Medicamentos de la Agencia de Registro de Medicamentos, Dispositivos Médicos y Productos Biocidas (Al. Jerozolimskie 181C, 02-222 Varsovia),
teléfono: 22 4921301,
fax: 22 4921309,
sitio web: https://smz.ezdrowie.gov.pl
Los efectos adversos también pueden notificarse al titular de la autorización de comercialización.
5. Cómo conservar Ig VENA
El medicamento debe conservarse en un lugar donde no pueda ser visto o alcanzado por los niños.
No debe utilizarse después de la fecha de caducidad impresa en la etiqueta de la ampolla y el paquete "EXP". La fecha de caducidad es el último día del mes indicado.
Conservar en refrigerador (2 °C - 8 °C).
Antes de su uso y dentro del período de validez, el medicamento puede conservarse a temperatura ambiente, no superior a 25 °C, durante un máximo de 6 meses consecutivos. Después de este tiempo, el medicamento debe ser eliminado. En ningún caso se puede volver a colocar el medicamento en el refrigerador si se ha conservado a temperatura ambiente. En el paquete debe anotarse la fecha inicial de conservación a temperatura ambiente.
Después de abrir la ampolla, el contenido debe utilizarse de inmediato.
Las ampollas deben conservarse en el paquete exterior. No congelar. No debe utilizarse este medicamento si se observa que la solución se ha vuelto turbia, ha cambiado de color o tiene un precipitado visible.
No debe tirarse el medicamento por el desagüe ni a los contenedores de basura domésticos. Debe preguntar a su farmacéutico cómo eliminar los medicamentos que ya no se necesitan. Este comportamiento ayudará a proteger el medio ambiente.
6. Contenido del paquete y otra información
Qué contiene Ig VENA?
El principio activo de este medicamento es la inmunoglobulina humana normal.
1 ml de solución contiene 50 mg de inmunoglobulina humana normal.
La solución contiene proteína humana 50 g/l, de la cual al menos 95% es IgG (inmunoglobulina G).
La distribución de las subclases de IgG es la siguiente:
IgG1
62,1%
IgG2
34,8%
IgG3
2,5%
IgG4
0,6%
El contenido máximo de IgA es de 50 microgramos/ml.
El medicamento se ha producido a partir de plasma de donantes de sangre.
Los demás componentes del medicamento son maltosa, agua para inyección.
Cómo se presenta Ig VENA y qué contiene el paquete?
La solución para infusión Ig VENA está disponible en ampollas individuales de 50 ml, 100 ml o 200 ml con un soporte instalado (ampolla + soporte). La solución es transparente o ligeramente opalescente, incolora o de color amarillo claro.
Tamaños de los paquetes:
Paquetes individuales:
1 ampolla que contiene 2,5 g/50 ml
1 ampolla que contiene 5 g/100 ml
1 ampolla que contiene 10 g/200 ml
Paquetes múltiples:
Paquete múltiple que contiene 2 paquetes individuales de 1 ampolla 10 g/200 ml
Paquete múltiple que contiene 3 paquetes individuales de 1 ampolla 10 g/200 ml.
No todos los tamaños de paquete pueden estar disponibles en el mercado.
Título de la autorización de comercialización:
Kedrion S.p.A.
Loc. Ai Conti, 55051 Castelvecchio Pascoli, Barga (Lucca), Italia
Fabricante:
Kedrion S.p.A.
55027 Bolognana, Gallicano (Lucca), Italia
Este medicamento está autorizado en los estados miembros del Espacio Económico Europeo bajo los siguientes nombres:
| Austria | Ig Vena 50g/l solución para infusión |
| Alemania | Ig Vena 50 g/l solución para infusión |
| Grecia | Ig VENA |
| Italia | IG VENA |
| Polonia | Ig VENA |
| Portugal | Ig Vena |
Para obtener información más detallada, debe consultar al representante local del titular de la autorización de comercialización:
MB&S Medical Business and Science, ul. Chełmska 30/34, 00-725 Varsovia
Teléfono/fax: 22 851 52 08
Fecha de la última revisión del prospecto: 11/2020
Información destinada exclusivamente a profesionales de la salud:
Instrucciones para la administración
- Antes de su uso, el producto Ig VENA debe alcanzar la temperatura ambiente o la temperatura corporal.
- Antes de su uso, la solución debe ser evaluada visualmente para detectar la presencia de partículas sólidas y decoloración. No debe utilizarse si la solución es turbia o tiene un precipitado.
- La inmunoglobulina humana normal debe administrarse por vía intravenosa con una velocidad inicial de infusión de 0,46 - 0,92 ml/kg/h (10 - 20 gotas por minuto) durante 20 - 30 minutos. En caso de reacción adversa, debe reducirse la velocidad de infusión o interrumpirse la administración. Si es bien tolerada, la velocidad de infusión puede aumentarse gradualmente hasta un máximo de 1,85 ml/kg/h (40 gotas por minuto).
- En pacientes con deficiencia primaria de la inmunidad que toleran una velocidad de infusión de 0,92 ml/kg/h, puede aumentarse gradualmente la velocidad de administración cada 20-30 minutos hasta 2 ml/kg/h, 4 ml/kg/h y como máximo 6 ml/kg/h, siempre y cuando el paciente la tolere bien. En general, la dosificación y la velocidad de infusión deben adaptarse individualmente a las necesidades del paciente. Dependiendo del peso corporal del paciente, la dosificación y la aparición de reacciones adversas, la velocidad de infusión máxima puede no alcanzarse. En caso de reacciones adversas, debe interrumpirse inmediatamente la infusión y luego reanudarse con una velocidad adecuada para el paciente.
Poblaciones especiales
En niños y adolescentes (0-18 años) y en personas de edad avanzada (>64 años), la velocidad inicial de administración debe ser de 0,46 – 0,92 ml/kg/h (10 – 20 gotas por minuto) durante 20 - 30 minutos. Si es bien tolerada, después de considerar la condición clínica del paciente, la velocidad puede aumentarse gradualmente hasta un máximo de 1,85 ml/kg/h (40 gotas por minuto).
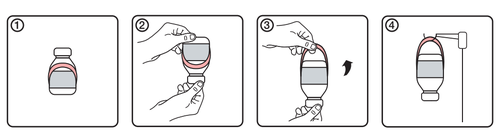
Instrucciones para el uso del soporte
- 1.Aspecto inicial de la ampolla con la etiqueta del soporte
- 2.Girar la ampolla hacia abajo
- 3.Crear el soporte desenrollándolo de la etiqueta
- 4. Colgar la ampolla en el soporte para infusión
Precauciones
Algunos efectos adversos graves pueden estar relacionados con la velocidad de infusión.
Se pueden evitar complicaciones potenciales asegurando que:
- los pacientes no sean alérgicos a la inmunoglobulina humana normal, mediante la administración inicial lenta del producto (velocidad de administración 0,46 - 0,92 ml/kg/h);
- los pacientes estén monitorizados y observados cuidadosamente durante la infusión para detectar reacciones adversas. Los pacientes que reciben inmunoglobulina humana normal por primera vez, los pacientes que han recibido previamente un producto IVIg diferente o en caso de una pausa prolongada desde la última infusión, deben ser monitorizados durante la primera infusión y durante la primera hora después de la primera infusión, con el fin de detectar signos de posibles reacciones adversas. Los demás pacientes deben ser observados durante al menos 20 minutos después de la infusión.
En todos los pacientes, la administración intravenosa de Ig requiere:
- hidratación adecuada antes de iniciar la infusión de Ig
- monitorización de la diuresis
- monitorización de los niveles de creatinina en suero
- evitar el uso concomitante de diuréticos de asa. En caso de una reacción adversa, debe reducirse la velocidad de administración o interrumpirse la administración de inmunoglobulina. El tratamiento depende del tipo y la gravedad de la reacción adversa. En caso de anafilaxia, debe procederse según los estándares médicos actuales para el tratamiento de la anafilaxia.
Reacción a la infusión
Algunos efectos adversos (por ejemplo, dolor de cabeza, escalofríos, fiebre, dolor muscular, respiración sibilante, taquicardia, dolor lumbar y náuseas) pueden estar relacionados con la velocidad de la infusión.
Debe seguir estrictamente la velocidad de infusión recomendada. Los pacientes deben ser monitorizados y observados cuidadosamente durante la infusión debido al riesgo de efectos adversos.
Algunas reacciones adversas pueden ocurrir con más frecuencia:
- en pacientes que reciben inmunoglobulina humana normal por primera vez, o en casos raros, cuando se cambia el producto de inmunoglobulina humana normal por otro o cuando se administra el producto después de una pausa prolongada
- en pacientes con infección no tratada o con una condición inflamatoria crónica.
Niños y adolescentes
No hay recomendaciones especiales para medidas de precaución o monitorización en niños y adolescentes. No se espera que haya diferencias entre los niños y adolescentes (de 0 a 18 años).
Enfermedad tromboembólica
Existen pruebas clínicas que sugieren una relación entre la administración intravenosa de Ig y casos de trombosis, como infarto de miocardio, accidente cerebrovascular, trombosis pulmonar o trombosis venosa profunda, que se consideran relacionados con el aumento relativo de la viscosidad sanguínea después de la administración de inmunoglobulina en pacientes con factores de riesgo.
Debe tenerse cuidado al prescribir y administrar el producto a pacientes obesos y a pacientes con riesgo de trombosis (como edad avanzada, hipertensión, diabetes, enfermedades vasculares o trombosis en la historia, trastornos adquiridos o congénitos de la coagulación, inmovilización prolongada, pacientes con hipovolemia grave, pacientes con enfermedades que se caracterizan por un aumento de la viscosidad sanguínea).
En pacientes con riesgo de trombosis, las inmunoglobulinas para administración intravenosa deben administrarse con la velocidad de infusión mínima y con la dosis más baja posible.
Insuficiencia renal aguda
Se han registrado casos de insuficiencia renal aguda en pacientes tratados con inmunoglobulinas para administración intravenosa. En la mayoría de estos casos, se identificaron factores de riesgo, como insuficiencia renal preexistente, diabetes, reducción grave del volumen sanguíneo, sobrepeso, uso concomitante de productos con propiedades nefrotóxicas o edad superior a 65 años.
Debe evaluarse la función renal antes de la administración de la infusión de IVIg y nuevamente en intervalos adecuados, especialmente en pacientes con un riesgo potencialmente aumentado de insuficiencia renal aguda. En pacientes con riesgo de insuficiencia renal aguda, las inmunoglobulinas intravenosas deben administrarse con la velocidad de infusión mínima y con la dosis más baja posible.
En caso de daño renal, debe considerarse la interrupción de la administración de Ig.
Meningitis aséptica
Se han registrado casos de síndrome de meningitis aséptica (AMS, por sus siglas en inglés) durante el tratamiento con inmunoglobulinas intravenosas. El síndrome generalmente comienza dentro de las primeras horas o 2 días después de la administración de IVIg.
En los análisis del líquido cefalorraquídeo, a menudo se encuentra pleocitosis con varios miles de células por mm3, principalmente granulocitos, y se encuentran niveles elevados de proteínas.
La meningitis aséptica puede ocurrir con más frecuencia en relación con el tratamiento con dosis altas de IVIg (2 g/kg).
Los pacientes con síntomas objetivos y subjetivos deben someterse a un examen neurológico detallado, incluido el análisis del líquido cefalorraquídeo, para excluir otras causas de meningitis.
La interrupción del tratamiento con IVIg provocó la remisión de la meningitis aséptica en el transcurso de varios días sin secuelas.
Anemia hemolítica
Los productos de inmunoglobulina intravenosa pueden contener anticuerpos contra los grupos sanguíneos, que pueden actuar como hemolizinas y inducir la cubierta de los glóbulos rojos con inmunoglobulina in vivo, lo que provoca una reacción directa positiva a la prueba de anticuerpos (prueba de Coombs) y, raramente, hemólisis.
La anemia hemolítica puede desarrollarse durante el tratamiento con IVIg como resultado de una secuestación aumentada de los glóbulos rojos. Los pacientes que reciben inmunoglobulina intravenosa deben ser monitorizados para detectar signos clínicos de hemólisis.
Neutropenia/leucopenia
Se han notificado casos de reducción transitoria del recuento de neutrófilos y/o episodios de neutropenia después del tratamiento con IVIg, a veces graves. Generalmente ocurren dentro de las primeras horas o días después de la administración de IVIg y se resuelven espontáneamente en el transcurso de 7 a 14 días.
Lesión pulmonar aguda relacionada con la transfusión (TRALI, por sus siglas en inglés)
Se han notificado casos de lesión pulmonar aguda no cardiogénica (TRALI, por sus siglas en inglés) en pacientes que reciben productos de IVIg. El TRALI se caracteriza por hipoxia grave, insuficiencia respiratoria, dificultad para respirar, cianosis, fiebre y disnea. Los síntomas del TRALI generalmente ocurren dentro de las 6 horas después de la administración del producto de IVIg, a menudo dentro de 1-2 horas. Por lo tanto, es necesario monitorizar a los pacientes; en caso de reacciones adversas respiratorias, debe interrumpirse inmediatamente la infusión de IVIg. La aparición de TRALI puede ser mortal, requiere tratamiento inmediato en la unidad de cuidados intensivos.
Este medicamento contiene 100 mg de maltosa por ml como excipiente. La presencia de maltosa en la sangre puede afectar el resultado de la prueba de glucosa, dando una lectura falsamente elevada de la concentración de glucosa en sangre, lo que puede llevar a una administración inadeuada de insulina, lo que puede provocar una hipoglucemia peligrosa para la vida. Además, los casos de hipoglucemia real pueden pasar desapercibidos si el estado de hipoglucemia está enmascarado por lecturas falsamente elevadas de la concentración de glucosa. En este contexto, al administrar Ig VENA o otros medicamentos para administración parenteral que contengan maltosa, la medición de la concentración de glucosa en sangre debe realizarse mediante un método específico para la glucosa. Debe leer cuidadosamente la información proporcionada con las tiras reactivas para la prueba de glucosa en sangre, incluida la información sobre las tiras reactivas, para determinar si se pueden utilizar con medicamentos para administración parenteral que contengan maltosa. En caso de duda, debe ponerse en contacto con el fabricante del dispositivo para determinar si se pueden utilizar con medicamentos para administración parenteral que contengan maltosa.
Dosificación
El tratamiento de reemplazo debe ser iniciado y supervisado por un médico especialista en el tratamiento de la deficiencia de inmunidad.
La dosis y el esquema de dosificación dependen de las indicaciones. La dosis debe ser determinada individualmente para cada paciente en función de la respuesta clínica. La dosis basada en el peso corporal puede requerir ajustes en pacientes con bajo peso o sobrepeso.
Los siguientes esquemas de dosificación se proporcionan como una guía.
Tratamiento de reemplazo en síndromes de deficiencia de inmunidad primaria
La dosis debe ser determinada para alcanzar un nivel de IgG (medido antes de la siguiente infusión) de al menos 6 g/l o dentro del rango normal para la edad de la población. Desde el inicio del tratamiento hasta la normalización de los niveles, se necesitan de 3 a 6 meses (nivel estable de IgG). Se recomienda una dosis inicial de 0,4-0,8 g/kg administrada en una sola dosis, y luego al menos 0,2 g/kg administrada cada 3-4 semanas. La dosis requerida para alcanzar un nivel mínimo de IgG de 6 g/l es de 0,2-0,8 g/kg/mes. Después de alcanzar un estado estable, los intervalos entre infusiones son de 3-4 semanas. Debe determinarse y evaluarse el nivel de inmunoglobulina en relación con la frecuencia de infecciones. Para reducir la frecuencia de infecciones bacterianas, puede ser necesario aumentar la dosis para alcanzar un nivel más alto.
Deficiencias de inmunidad secundarias
La dosis recomendada es de 0,2-0,4 g/kg cada 3-4 semanas.
Debe determinarse y evaluarse el nivel mínimo de IgG en relación con la frecuencia de infecciones. La dosis debe ajustarse según sea necesario para lograr la protección adecuada contra las infecciones; puede ser necesario aumentar la dosis en pacientes con infecciones persistentes; la reducción de la dosis puede considerarse cuando el paciente no tenga infecciones.
Purpura trombocitopénica idiopática
Dos esquemas de tratamiento alternativos:
- dosis de 0,8-1,0 g/kg en el primer día; la dosis puede repetirse una vez en un período de 3 días
- 0,4 g/kg por día durante 2-5 días. El tratamiento puede repetirse si ocurre una recaída de la enfermedad.
Síndrome de Guillain-Barré
0,4 g/kg/día durante más de 5 días (puede repetirse la dosis en caso de recaída).
Enfermedad de Kawasaki
Debe administrarse una dosis única de 2,0 g/kg. Los pacientes deben recibir ácido acetilsalicílico al mismo tiempo.
Polieneuropatía demielinizante crónica inflamatoria (CIDP)
Dosis inicial: 2 g/kg durante 2-5 días consecutivos.
Dosis de mantenimiento: 1 g/kg durante 1-2 días consecutivos cada 3 semanas.
La eficacia del tratamiento debe evaluarse después de cada ciclo; si se observa falta de eficacia después de 6 meses, el tratamiento debe suspenderse.
Si la terapia es eficaz, el médico debe tomar la decisión de tratamiento a largo plazo teniendo en cuenta las reacciones del paciente y la respuesta al tratamiento de mantenimiento. La dosis y los intervalos de tratamiento pueden requerir ajustes según el curso individual de la enfermedad.
Neuropatía motora multifocal (MMN)
Dosis inicial: 2 g/kg administrada durante 2-5 días consecutivos.
Dosis de mantenimiento: 1 g/kg cada 2-4 semanas o 2 g/kg cada 4-8 semanas.
La eficacia del tratamiento debe evaluarse después de cada ciclo; si se observa falta de eficacia después de 6 meses, el tratamiento debe suspenderse.
Si la terapia es eficaz, el médico debe tomar la decisión de tratamiento a largo plazo teniendo en cuenta las reacciones del paciente y la respuesta al tratamiento de mantenimiento. La dosis y los intervalos de tratamiento pueden requerir ajustes según el curso individual de la enfermedad.
La dosis recomendada se presenta en la siguiente tabla:
| Indicaciones | Dosis | Frecuencia de infusiones |
| Tratamiento de reemplazo | ||
| Síndromes de deficiencia de inmunidad primaria | dosis inicial: 0,4-0,8 g/kg, dosis de mantenimiento: 0,2-0,8 g/kg | cada 3-4 semanas |
| Deficiencias de inmunidad secundarias | 0,2-0,4 g/kg | cada 3-4 semanas |
| Tratamiento inmunomodulador | ||
| Purpura trombocitopénica idiopática | 0,8-1 g/kg o 0,4 g/kg/día | en el primer día, posible repetición una vez en un período de 3 días, durante 2-5 días |
| Síndrome de Guillain-Barré | 0,4 g/kg/día | durante 5 días |
| Enfermedad de Kawasaki | 2 g/kg | en una dosis única en combinación con ácido acetilsalicílico |
| Polieneuropatía demielinizante crónica inflamatoria (CIDP) | dosis inicial: 2 g/kg, dosis de mantenimiento: 1 g/kg | en dosis divididas durante 2-5 días, cada 3 semanas durante 1-2 días |
| Neuropatía motora multifocal (MMN) | dosis de mantenimiento: 1 g/kg o 2 g/kg | cada 2-4 semanas o cada 4-8 semanas durante 2-5 días |
Uso en niños y adolescentes
La dosis en niños y adolescentes (0-18 años) no difiere de la utilizada en adultos, ya que la dosis en las diferentes indicaciones se determina en función del peso corporal y el estado clínico del paciente, según lo anterior.
Pacientes con insuficiencia renal
No hay datos disponibles sobre la necesidad de ajustar la dosis.
Pacientes con insuficiencia hepática
No hay necesidad de ajustar la dosis, a menos que esté justificado clínicamente.
Pacientes ancianos
No hay necesidad de ajustar la dosis, a menos que esté justificado clínicamente.
CIDP
Debido a la rareza de la polineuropatía demielinizante crónica inflamatoria y, en consecuencia, al pequeño número de pacientes en general, la experiencia en el uso de inmunoglobulina intravenosa en niños con CIDP es limitada; por lo tanto, solo están disponibles datos de la literatura. Sin embargo, los datos publicados son consistentes y todos muestran que el tratamiento con IVIg en adultos y niños es igual de eficaz que en las indicaciones aprobadas hasta ahora.
- País de registro
- Principio activo
- Requiere recetaNo
- Fabricante
- ImportadorKedrion S.p.A.
- Esta información ha sido traducida con IA y es solo orientativa. No constituye asesoramiento médico. Consulta siempre con un médico antes de tomar cualquier medicamento.
- Alternativas a Ig VenaPrincipio activo: immunoglobulins, normal human, for intravascular adm.Fabricante: Baxalta Belgium Manufacturing S.A.Requiere recetaForma farmacéutica: Solución, 100 mg/mlPrincipio activo: immunoglobulins, normal human, for intravascular adm.Fabricante: Instituto Grifols S.A.Requiere recetaForma farmacéutica: Solución, 50 g/l (50 mg/ml)Principio activo: immunoglobulins, normal human, for intravascular adm.Fabricante: Biotest Pharma GmbHRequiere receta
Alternativas a Ig Vena en otros países
Las mejores alternativas con el mismo principio activo y efecto terapéutico.
Alternativa a Ig Vena en España
Alternativa a Ig Vena en Ucrania
Médicos online para Ig Vena
Consulta sobre dosis, efectos secundarios, interacciones, contraindicaciones y renovación de la receta de Ig Vena – sujeta a valoración médica y normativa local.









Mantente informado y ahorra en salud
Recibe consejos de salud, novedades de la plataforma y promociones exclusivas para suscriptores.

