

Anbinex
Consulta con un médico sobre la receta médica de Anbinex



Cómo usar Anbinex
HOJA DE INSTRUCCIONES DEL PACIENTE: INFORMACIÓN PARA EL USUARIO
Anbinex
50 UI/ml; 500 UI y 1000 UI
Polvo y disolvente para la preparación de una solución para infusión. Antitrombina III humana concentrada
Es importante leer el folleto antes de tomar el medicamento, ya que contiene información importante para el paciente
- Conservar este folleto para poder volver a leerlo si es necesario.
- En caso de dudas, consultar al médico, farmacéutico o enfermera
- Este medicamento ha sido recetado exclusivamente para una persona determinada. No debe ser entregado a otros. El medicamento puede ser perjudicial para otra persona, incluso si los síntomas de su enfermedad son los mismos.
- Si el paciente experimenta algún efecto adverso, incluidos todos los efectos adversos no mencionados en este folleto, debe informar al médico, farmacéutico o enfermera. Ver punto 4.
Índice del folleto:
- 1. Qué es Anbinex y para qué se utiliza
- 2. Información importante antes de tomar Anbinex
- 3. Cómo tomar Anbinex
- 4. Posibles efectos adversos
- 5. Cómo conservar Anbinex
- 6. Contenido del envase y otra información
1. Qué es Anbinex y para qué se utiliza
Anbinex es
un medicamento
antitrombótico,
pertenece
a la clase de medicamentos antitrombóticos parenterales.
Este medicamento se utiliza para tratar el déficit congénito de antitrombina, para prevenir la formación de trombosis venosas profundas en las extremidades inferiores y para prevenir las complicaciones tromboembólicas en otros vasos sanguíneos.
Si hay indicaciones para ello, también se administra durante procedimientos quirúrgicos y en el período periparto. En algunos casos, puede administrarse en combinación con heparina.
Anbinex también se utiliza para tratar el déficit adquirido de antitrombina.
2. Información importante antes de tomar Anbinex
Cuándo no tomar Anbinex
Si el paciente es alérgico a la antitrombina o a alguno de los demás componentes de este medicamento (enumerados en el punto 6).
Es importante leer la información importante al final de este punto.
Precauciones y advertencias
Antes de comenzar a tomar Anbinex, es importante discutirlo con el médico, farmacéutico o enfermera.
Al igual que con otros productos administrados por vía intravenosa, es posible que ocurran reacciones alérgicas. Durante la infusión, el paciente debe ser monitoreado de cerca debido al riesgo de efectos adversos. En caso de experimentar síntomas de reacción de hipersensibilidad, incluyendo erupciones, urticaria generalizada, sensación de opresión en el pecho, respiración sibilante (dificultad para respirar), hipotensión y síntomas de anafilaxia, debe informar inmediatamente al médico que lo está tratando.
En el proceso de fabricación de medicamentos a partir de sangre o plasma humanos, se siguen procedimientos específicos para prevenir la transmisión de infecciones a los pacientes que los reciben. Estos procedimientos incluyen:
- selección detallada de donantes de sangre y plasma, con el fin de excluir a los donantes que puedan ser una fuente de infección;
- análisis de cada donación y lote de plasma extraído para detectar la presencia de virus/agentes infecciosos;
- incorporación de etapas en el proceso de tratamiento del plasma, durante las cuales los virus pueden ser inactivados o eliminados.
A pesar de la implementación de estas medidas de precaución, no se puede descartar por completo la posibilidad de transmisión de infecciones si se administran medicamentos fabricados a partir de sangre o plasma humanos. Esto también se aplica a los virus y patógenos desconocidos o recientemente descubiertos.
Se considera que las medidas de precaución adoptadas son efectivas contra los virus con envoltura, como el virus de la inmunodeficiencia humana adquirida (VIH), el virus de la hepatitis B, el virus de la hepatitis C y el virus de la hepatitis A sin envoltura. Las medidas anteriores pueden tener un valor limitado en el caso de los virus sin envoltura, como el parvovirus B19.
La infección por parvovirus B19 puede ser particularmente grave en mujeres embarazadas (infección fetal) y en personas con sistema inmunitario debilitado o que padecen ciertos tipos de anemia (por ejemplo, anemia falciforme, anemia hemolítica).
En pacientes que reciben regularmente/múltiples veces antitrombina humana derivada de plasma, el médico puede recomendar la vacunación contra el virus de la hepatitis A y B.
Se recomienda encarecidamente que, en cada administración de Anbinex al paciente, se registre el nombre del paciente y el número de lote del producto, para poder vincular al paciente con el lote del medicamento.
Anbinex y otros medicamentos
Es importante informar al médico o farmacéutico sobre todos los medicamentos que el paciente está tomando actualmente o ha tomado recientemente, así como sobre los medicamentos que el paciente planea tomar.
La administración de antitrombina al mismo tiempo que dosis terapéuticas de heparina aumenta el riesgo de hemorragias. El efecto de la antitrombina se potencia significativamente con la heparina. La administración concomitante de heparina a pacientes con mayor riesgo de hemorragias debe ser monitoreada cuidadosamente desde el punto de vista clínico y biológico.
Embarazo y lactancia
Si la paciente está embarazada o en período de lactancia, sospecha que puede estar embarazada o planea tener un hijo, debe consultar a su médico o farmacéutico antes de tomar este medicamento.
Anbinex debe ser utilizado durante el embarazo y la lactancia solo si es estrictamente necesario. La decisión debe ser tomada después de considerar que, durante el embarazo, existe un mayor riesgo de desarrollar eventos tromboembólicos.
Conducción de vehículos y uso de máquinas
Anbinex no tiene o tiene un efecto mínimo en la capacidad para conducir vehículos y utilizar máquinas.
Anbinex contiene sodio
Anbinex 500 UI contiene 1,45 mmol (33,35 mg) de sodio en 10 ml.
Anbinex 1000 UI contiene 2,90 mmol (66,7 mg) de sodio en 20 ml
Es importante tener en cuenta a los pacientes que están en dietas con restricción de sal.
3. Cómo tomar Anbinex
El medicamento Anbinex para administración por infusión intravenosa debe ser preparado por un médico o enfermera.
Uso en niños y adolescentes
Debido a la falta de datos suficientes, no se recomienda el uso de Anbinex en niños menores de 6 años.
Frecuencia de administración
El médico determinará la frecuencia de administración de Anbinex y los intervalos entre dosis.
Duración del tratamiento.
El médico determinará la duración del tratamiento con Anbinex.
En caso de sobredosis de Anbinex
No se han notificado casos de sobredosis.
4. Posibles efectos adversos
Como cualquier medicamento, Anbinex puede causar efectos adversos, aunque no todos los pacientes los experimentarán.
En casos raros, se han observado aumento de la temperatura corporal y reacciones alérgicas o anafilácticas, como enrojecimiento de la cara, erupciones, aumento o disminución de la presión arterial, taquicardia (aceleración del ritmo cardíaco), escalofríos, respiración sibilante y edemas, así como reacciones generalizadas (incluyendo dolor en el pecho, fiebre, dolor de cabeza, náuseas y/o vómitos), que en algunos casos han llevado al desarrollo de una reacción anafiláctica grave (incluyendo shock).
En casos raros, se han observado aumento de la temperatura corporal.
Lista de efectos adversos en forma tabular.
La frecuencia de los efectos adversos se evaluó utilizando los siguientes criterios:
- muy frecuente (> 1/10),
- frecuente (> 1/100, <1>
- no muy frecuente (> 1/1000, <1>
- raros (> 1/10 000, <1>
- muy raros (<1>
- frecuencia desconocida (frecuencia no puede ser determinada a partir de los datos disponibles)
| Sistema de clasificación MedDRA (SOC) | Reacción adversa | Frecuencia de ocurrencia |
| Trastornos del sistema inmunológico | Reacciones alérgicas, hipersensibilidad | No muy frecuente |
| Trastornos psiquiátricos | Ansiedad | No muy frecuente |
| Trastornos del sistema nervioso | Dolores de cabeza, letargo | No muy frecuente |
| Trastornos del sistema cardiovascular | Taquicardia | No muy frecuente |
| Trastornos vasculares | Enrojecimiento de la cara, disminución de la presión arterial, shock | No muy frecuente |
| Trastornos del sistema respiratorio, torácico y mediastínico | Sensación de opresión en el pecho y sibilancias | No muy frecuente |
| Trastornos gastrointestinales | Náuseas, vómitos, | No muy frecuente |
| Trastornos de la piel y del tejido subcutáneo | Edema angioneurótico, urticaria generalizada, erupciones | No muy frecuente |
| Trastornos generales y condiciones en el lugar de administración | Dolor o sensación de quemazón en el lugar de administración, escalofríos | No muy frecuente |
| Fiebre | Raro |
Es importante informar al médico si ocurre alguno de estos síntomas.
Información sobre las medidas de seguridad para prevenir la transmisión de agentes infecciosos - ver punto 2.
Si ocurren efectos adversos, incluidos todos los efectos adversos no mencionados en el folleto, debe informar al médico, farmacéutico o enfermera.
Los efectos adversos pueden ser notificados directamente al Departamento de Vigilancia de Reacciones Adversas de Medicamentos de la Agencia de Registro de Productos Farmacéuticos, Dispositivos Médicos y Productos Biocidas:
Aleja Jerozolimskie 181C,
02-222 Varsovia,
Tel.: + 48 22 49 21 301,
Fax: + 48 22 49 21 309,
correo electrónico: [email protected]
Los efectos adversos también pueden ser notificados al titular de la autorización de comercialización.
Gracias a la notificación de los efectos adversos, se podrán recopilar más información sobre la seguridad del medicamento.
5. Cómo conservar Anbinex
El medicamento debe conservarse en un lugar donde no sea visible y no esté al alcance de los niños.
No debe utilizarse después de la fecha de caducidad (EXP) que figura en la etiqueta. La fecha de caducidad es el último día del mes indicado
No debe conservarse a una temperatura superior a 30°C. No debe congelarse.
Después de la reconstitución:
Los estudios de estabilidad indican un período de validez de hasta 12 horas a 25°C. Desde el punto de vista microbiológico, el producto debe ser utilizado de inmediato. Si después de la reconstitución el producto no se ha utilizado, puede conservarse durante un máximo de 24 horas a una temperatura de 2°C - 8°C, pero solo si el usuario asume la responsabilidad y la preparación de la solución se ha realizado de acuerdo con las normas de esterilidad.
La solución debe ser transparente y ligeramente opalescente.
No debe utilizarse si se observan partículas o sedimentos.
No debe desecharse por el desagüe o en contenedores de residuos domésticos. Debe preguntar al farmacéutico cómo eliminar los medicamentos que ya no se utilizan. Este procedimiento ayudará a proteger el medio ambiente.
6. Contenido del envase y otra información
Qué contiene Anbinex?
El principio activo del medicamento es la antitrombina humana.
El frasco con polvo contiene 500 UI o 1000 UI de antitrombina humana.
Después de la reconstitución, el producto contiene 50 UI/ml (500 UI/10 ml o 1000 UI/20 ml) de antitrombina humana.
Los demás componentes son: D-mannitol, cloruro de sodio y citrato de sodio.
La jeringa ampolla contiene 10 ml o 20 ml de agua para inyección.
Para obtener más información sobre los componentes, ver punto 2.
Cómo se presenta Anbinex y qué contiene el envase?
El envase contiene un frasco con una sustancia blanca, higroscópica, quebradiza o en polvo, y una jeringa ampolla con agua para inyección.
Cada envase de Anbinex 500 UI contiene un frasco con 500 UI de antitrombina humana (polvo para la preparación de una solución para infusión) y 1 jeringa ampolla con 10 ml de agua para inyección (disolvente).
Cada envase de Anbinex 1000 UI contiene un frasco con 1000 UI de antitrombina humana (polvo para la preparación de una solución para infusión) y 1 jeringa ampolla con 20 ml de agua para inyección (disolvente).
Con cada envase de Anbinex se proporciona un conjunto para la preparación de la solución que incluye un conector de sujeción para el frasco y un microfiltro.
Titular de la autorización de comercialización y fabricante
Instituto Grifols, S.A.
Polígono Levante, c/Can Guasch, 2Parets del Vallès
08150 Barcelona, ESPAÑA.
Para obtener información más detallada, debe dirigirse al representante del titular de la autorización de comercialización.
Grifols Polska Sp. z o. o.
Ul. Grzybowska 87, 00-844 Varsovia
Tel.: + 48 22 5040641
Fecha de la última actualización del folleto:
----------------------------------------------------------------------------------------------------------------
Informaciones destinadas exclusivamente al personal médico especializado:
En el déficit congénito, la dosis y la duración del tratamiento deben ser adaptadas individualmente para cada paciente, dependiendo del resultado de la historia familiar que tenga en cuenta los casos de eventos tromboembólicos, los factores de riesgo clínico actuales y los resultados de los análisis de laboratorio.
La dosis y la duración del tratamiento de reemplazo en el déficit adquirido de antitrombina dependen del nivel de antitrombina en el suero, la presencia de síntomas que indiquen un consumo acelerado, la enfermedad subyacente y la gravedad de los síntomas clínicos. El tamaño de las dosis y la frecuencia de administración deben adaptarse siempre individualmente para cada paciente, dependiendo de los efectos clínicos.
La dosis administrada de antitrombina se expresa en unidades internacionales (UI), de acuerdo con las normas actuales de la OMS. La actividad de la antitrombina en el suero puede expresarse en porcentajes (en relación con la actividad en suero normal) o en unidades internacionales (de acuerdo con el estándar internacional para la antitrombina del suero).
Una unidad internacional (UI) de actividad de la antitrombina equivale a la cantidad media de antitrombina en 1 ml de suero humano normal. El cálculo de la dosis necesaria de antitrombina se basa en la observación empírica de que la administración de 1 UI de antitrombina por kg de peso corporal aumenta la actividad de la antitrombina en el suero en aproximadamente 1,1% a 1,6%.
La dosis inicial se calcula según la siguiente fórmula:
Número de unidades requeridas = peso corporal (kg) x (100 – actividad inicial de antitrombina (en porcentaje) x 0,8
En la fase inicial del tratamiento, debe determinarse el nivel deseado de actividad de la antitrombina en función de la situación clínica. Después de determinar las indicaciones para el uso de la antitrombina, debe administrarse una dosis que permita alcanzar el nivel deseado de actividad de la antitrombina y, posteriormente, mantener su nivel para garantizar la eficacia del tratamiento.
La dosis debe calcularse y monitorearse en función de los análisis de laboratorio de la actividad de la antitrombina en el suero. Los análisis deben realizarse al menos dos veces al día, y cuando el estado del paciente se estabilice, una vez al día; siempre justo antes de la próxima administración del medicamento. Es importante tener en cuenta que, en el caso de estados clínicos graves, como el síndrome de coagulación intravascular diseminada, el tiempo de vida de la antitrombina puede ser significativamente más corto. La corrección del tamaño de la dosis debe realizarse teniendo en cuenta tanto la velocidad de consumo de la antitrombina determinada por los análisis de laboratorio como el curso clínico. La actividad de la antitrombina debe mantenerse por encima del 80% de la norma durante todo el período de tratamiento o adaptarse adecuadamente cuando los síntomas clínicos indiquen que otro nivel puede ser más eficaz.
En el tratamiento del déficit congénito, la dosis inicial es de 30-50 UI/kg de peso corporal.
Posteriormente, el tamaño de la dosis, la frecuencia de administración y la duración del tratamiento dependen de la respuesta biológica en la situación clínica dada.
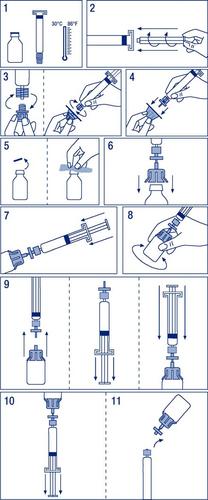
Instrucciones para la preparación del medicamento para su uso
- 1. Calentar los frascos a una temperatura no superior a 30°C (dibujo n.º 1).
- 2. Fijar el émbolo en la jeringa ampolla con el disolvente (dibujo n.º 2).
- 3. Retirar el filtro de su envase. Quitar la cubierta de plástico del extremo de la jeringa ampolla y fijar el filtro (dibujo n.º 3).
- 4. Retirar el conector de sujeción del frasco y conectar la jeringa ampolla con el filtro (dibujo n.º 4).
- 5. Quitar la cubierta de plástico del frasco y desinfectar el tapón de goma expuesto con un agente desinfectante (dibujo n.º 5).
- 6. Perforar el tapón del frasco con la aguja del conector (dibujo n.º 6).
- 7. Inyectar todo el disolvente en el frasco (dibujo n.º 7).
- 8. Agitar suavemente el frasco hasta que el polvo se disuelva (dibujo n.º 8).
- 9. Desconectar la jeringa ampolla con el filtro del frasco con el conector. Tirar del émbolo para permitir que entre aire en una cantidad equivalente al volumen del disolvente. Volver a conectar la jeringa con el filtro adjunto al frasco con el conector (dibujo n.º 9).
- 10. Invertir el frasco y aspirar la solución en la jeringa ampolla (dibujo n.º 10).
- 11. Desconectar la jeringa ampolla del filtro y del frasco, y administrar lentamente por vía intravenosa a una velocidad no superior a 0,08 ml/kg/min (dibujo n.º 11).
No dejar el producto sin usar para su uso posterior.
No reutilizar el conjunto de administración.
Al usar el conjunto de infusión, debe comprobarse su compatibilidad con la jeringa ampolla. Debe utilizarse un adaptador adecuado para garantizar la administración correcta del producto.
- País de registro
- Principio activo
- Requiere recetaNo
- Fabricante
- ImportadorInstituto Grifols, S.A.
- Esta información ha sido traducida con IA y es solo orientativa. No constituye asesoramiento médico. Consulta siempre con un médico antes de tomar cualquier medicamento.
- Alternativas a AnbinexForma farmacéutica: Polvo, 500 UIPrincipio activo: Antitrombina iiiFabricante: Instituto Grifols S.A.No requiere recetaForma farmacéutica: Polvo, 50 UI/ml; 500 UIPrincipio activo: Antitrombina iiiFabricante: Takeda Manufacturing Austria AGNo requiere recetaForma farmacéutica: Polvo, 50 UI/mlPrincipio activo: Antitrombina iiiFabricante: Takeda Manufacturing Austria AGNo requiere receta
Alternativas a Anbinex en otros países
Las mejores alternativas con el mismo principio activo y efecto terapéutico.
Alternativa a Anbinex en Ucrania
Alternativa a Anbinex en España
Médicos online para Anbinex
Consulta sobre dosis, efectos secundarios, interacciones, contraindicaciones y renovación de la receta de Anbinex – sujeta a valoración médica y normativa local.









Mantente informado y ahorra en salud
Recibe consejos de salud, novedades de la plataforma y promociones exclusivas para suscriptores.

