

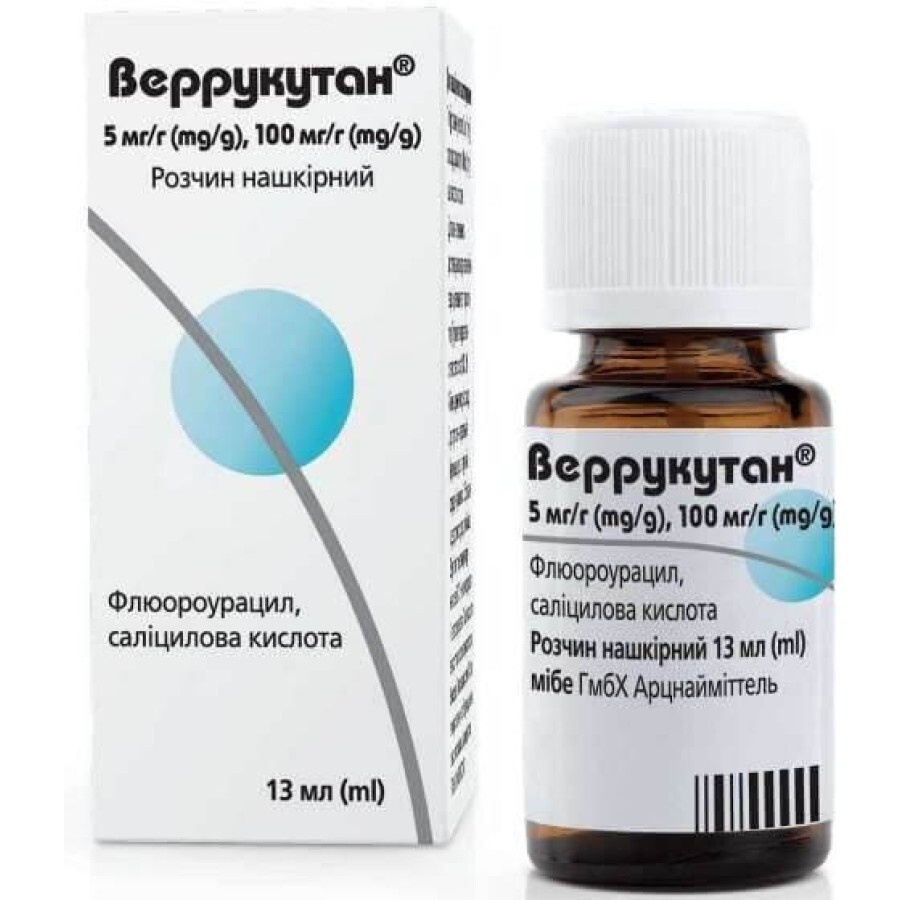
VERRUKUTAN
Pergunte a um médico sobre a prescrição de VERRUKUTAN



Como usar VERRUKUTAN
INSTRUÇÕES para uso médico do medicamento JAKAVI (JAKAVI®)
COMPOSIÇÃO
substância ativa: ruxolitinibe (ruxolitinib); 1 comprimido contém 5 mg, 15 mg ou 20 mg de ruxolitinibe (na forma de fosfato); excipientes: monoidratado de lactose, celulose microcristalina, glicolato de sódio (tipo A), hidroxipropilcelulose, povidona, dióxido de silício coloidal anidro, estearato de magnésio.
FORMA FARMACÊUTICA
Comprimidos.
PROPRIEDADES FÍSICO-QUÍMICAS
comprimidos de 5 mg: redondos, curvos, de cor branca a quase branca, comprimidos com aproximadamente 7,5 mm de diâmetro com gravação "NVR" em um lado e "L5" no outro; comprimidos de 15 mg: ovais, curvos, de cor branca a quase branca, comprimidos com dimensões de aproximadamente 15,0 x 7,0 mm com gravação "NVR" em um lado e "L15" no outro; comprimidos de 20 mg: alongados, curvos, de cor branca a quase branca, comprimidos com dimensões de aproximadamente 16,5 x 7,4 mm com gravação "NVR" em um lado e "L20" no outro.
GRUPO FARMACOTERAPÊUTICO
Agentes antineoplásicos. Inibidores de proteína quinase. Inibidores de janus quinase (JAK).
CÓDIGO ATC
L01E J01.
PROPRIEDADES FARMACOLÓGICAS
FARMACODINÂMICA
O ruxolitinibe é um inibidor seletivo de janus quinases (JAKs) JAK1 e JAK2 (valores de IC50 para as enzimas JAK1 e JAK2 são de 3,3 nM e 2,8 nM, respectivamente). Essas enzimas são intermediárias na transmissão de sinais de uma série de citocinas e fatores de crescimento que são importantes para a hematopoese e o sistema imunológico. A mielofibrose (MF) e a policitemia vera (PV) são neoplasias mieloproliferativas que, como se sabe, estão associadas a uma regulação anormal na transmissão de sinais que envolve JAK1 e JAK2. A base da regulação anormal é a presença de níveis elevados de citocinas circulantes que ativam a via JAK-STAT-mutações, nas quais o produto de expressão gênica mutante adquire novas funções patológicas, como a mutação JAK2V617F, e também levam à exclusão de mecanismos reguladores negativos. Em pacientes com MF, a regulação anormal na transmissão de sinais que envolve JAK é observada independentemente do status de mutação JAK2V617F. Mutações ativadoras de JAK2 (V617F ou exão 12) são observadas em mais de 95% dos pacientes com PV. O ruxolitinibe inibe a transmissão de sinais da via JAK-STAT e a proliferação de células em modelos celulares de neoplasias hematológicas dependentes de citocinas, bem como em células Ba/F3 que expressam a proteína JAK2V617F mutante com um IC50 na faixa de 80-320 nM.
EFETOS FARMACODINÂMICOS
O ruxolitinibe inibe a fosforilação induzida por citocinas de STAT3 no sangue total de voluntários saudáveis e pacientes com MF e PV. A administração de ruxolitinibe leva a uma inibição máxima da fosforilação de STAT3 dentro de 2 horas após a administração do medicamento, com um retorno aos níveis basais de fosforilação dentro de 8 horas, tanto em voluntários saudáveis quanto em pacientes com MF, o que sugere a ausência de acúmulo do medicamento original ou de metabólitos ativos. Os níveis elevados de marcadores de inflamação associados à ocorrência de sintomas sistêmicos, como o fator de necrose tumoral alfa (FNT-α), interleucina-6 (IL-6) e proteína C-reativa (PCR), em pacientes com MF, diminuíram após o tratamento com ruxolitinibe. Com o passar do tempo, em pacientes com MF, não se desenvolveu resistência aos efeitos farmacodinâmicos do tratamento com ruxolitinibe. Da mesma forma, em pacientes com PV, observou-se um aumento nos níveis basais de marcadores de inflamação, e esses níveis diminuíram após o tratamento com ruxolitinibe. Ao examinar a duração do intervalo QT em voluntários saudáveis, não se detectou nenhum sinal de prolongamento do QT/QTc com a administração de ruxolitinibe em doses únicas que excediam as doses terapêuticas (até 200 mg), o que sugere que o ruxolitinibe não tem efeito sobre a repolarização cardíaca.
FARMACOCINÉTICA
ABSORÇÃO
O ruxolitinibe pertence à classe 1 do Sistema de Classificação Biofarmacêutica (BCS) com alta permeabilidade, alta solubilidade e dissolução rápida. Durante os estudos clínicos, verificou-se que o ruxolitinibe é rapidamente absorvido após a administração oral, atingindo a concentração máxima no plasma dentro de aproximadamente 1 hora após a dose. Com base nos dados do estudo de balanço de massa corporal em humanos, a absorção do ruxolitinibe após a administração oral é de 95% ou mais. A Cmax e a exposição total (AUC) do ruxolitinibe aumentam proporcionalmente com o aumento da dose única na faixa de 5 a 200 mg. Não se observaram mudanças clinicamente significativas na farmacocinética do ruxolitinibe após a administração com alimentos ricos em gordura. Quando administrado com alimentos ricos em gordura, a Cmax média diminuiu moderadamente (24%), enquanto a AUC média permaneceu quase inalterada (aumento de 4%).
DISTRIBUIÇÃO
Em pacientes com MF e PV, o volume de distribuição no estado de equilíbrio é de aproximadamente 75 litros. A ligação do ruxolitinibe às proteínas plasmáticas é de aproximadamente 97% em condições in vitro. Um estudo de autorradiografia em todo o organismo em ratos mostrou que o ruxolitinibe não atravessa a barreira hematoencefálica.
BIOTRANSFORMAÇÃO
O ruxolitinibe é metabolizado principalmente pelo CYP3A4 (> 50%) com participação adicional do CYP2C9. A substância original é a forma dominante no plasma humano, representando cerca de 60% do material relacionado ao medicamento no plasma. Dois metabólitos principais e ativos foram detectados no plasma, representando 25% e 11% da AUC da substância original. Esses metabólitos têm de 1/2 a 1/5 da atividade farmacológica relacionada à JAK da substância original. No geral, a soma de todos os metabólitos ativos contribui até 18% da farmacodinâmica total do ruxolitinibe. Em concentrações clinicamente relevantes, o ruxolitinibe não inibe a atividade das enzimas CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ou CYP3A4 e não é um indutor potente das enzimas CYP1A2, CYP2B6 ou CYP3A4 com base nos resultados dos estudos in vitro. Os dados obtidos in vitro sugerem que o ruxolitinibe pode inibir a atividade da glicoproteína P e da proteína de resistência ao câncer de mama (BCRP).
ELIMINAÇÃO
O ruxolitinibe é eliminado principalmente por metabolismo. O período de meia-vida do ruxolitinibe é de aproximadamente 3 horas. Após a administração de uma dose única de [14C]-ruxolitinibe em voluntários saudáveis, a eliminação ocorreu principalmente por metabolismo, com 74% do composto radioativamente marcado eliminado na urina e 22% nas fezes. A substância original inalterada representou menos de 1% do composto radioativamente marcado eliminado.
LINEARIDADE/NÃO LINEARIDADE
A proporcionalidade da dose foi demonstrada nos estudos de dose única e dose repetida.
GRUPOS ESPECIAIS DE PACIENTES
INFLUÊNCIA DA ÁREA DE SUPERFÍCIE CORPORAL, IDADE, SEXO OU RAÇA
Nos estudos com voluntários saudáveis de diferentes sexos e raças, não se observaram diferenças significativas na farmacocinética do ruxolitinibe. Durante a avaliação farmacocinética populacional em pacientes com MF, não se encontrou nenhuma dependência aparente da depuração oral com a idade ou raça do paciente. A depuração oral prevista foi de 17,7 L/h em mulheres e 22,1 L/h em homens, com variabilidade de 39%. Em pacientes com PV, a depuração foi de 12,7 L/h, com variabilidade de 42%. Durante a avaliação farmacocinética populacional em pacientes com PV, não se encontrou nenhuma dependência da depuração oral com o sexo, idade ou raça do paciente. A depuração foi de 10,4 L/h em pacientes com Síndrome Mielodisplásica (SMD) aguda e 7,8 L/h em pacientes com SMD crônica, com variabilidade interindividuais de 49%. Durante a avaliação farmacocinética populacional em pacientes com SMD, não se encontrou nenhuma dependência da depuração oral com o sexo, idade ou raça do paciente. A exposição aumentou em pacientes com SMD e baixa área de superfície corporal (ASC). Em pacientes com ASC de 1 m2, 1,25 m2 e 1,5 m2, a exposição média prevista (AUC) foi 31%, 22% e 12% maior, respectivamente, do que em um adulto típico (1,79 m2).
POPLAÇÃO PEDIÁTRICA
A farmacocinética do medicamento JAKAVI em crianças (menores de 18 anos) com MF e PV não foi estabelecida. O perfil farmacocinético observado em crianças com SMD aguda ou crônica foi comparável ao da população geral de pacientes.
O ruxolitinibe ainda não foi estudado em crianças menores de 12 anos com SMD aguda ou crônica.
DISFUNÇÃO RENAL
A função renal foi avaliada usando a fórmula de depuração de creatinina (MDRD) e a taxa de filtração glomerular (TFG). Após a administração de uma dose única de 25 mg de ruxolitinibe, a exposição foi semelhante em pacientes com diferentes graus de disfunção renal e em indivíduos com função renal normal. No entanto, os valores de AUC dos metabólitos do ruxolitinibe no plasma aumentaram com a gravidade da disfunção renal e foram mais pronunciados em pacientes com disfunção renal grave. É desconhecido se o aumento da exposição aos metabólitos tem implicações de segurança. A alteração da dose do medicamento é recomendada para pacientes com disfunção renal grave e estágio terminal de doença renal (ver seção "Posologia e Administração"). A administração do medicamento apenas nos dias de diálise reduz a exposição aos metabólitos e o efeito farmacodinâmico, especialmente nos dias entre as sessões de diálise.
DISFUNÇÃO HEPÁTICA
Após a administração de uma dose única de 25 mg de ruxolitinibe, o valor médio de AUC do ruxolitinibe aumentou em pacientes com disfunção hepática leve, moderada e grave em 87%, 28% e 65%, respectivamente, em comparação com pacientes com função hepática normal. Não se encontrou uma correlação clara entre a AUC do medicamento e o grau de disfunção hepática com base nos escores da escala de Child-Pugh. O período de meia-vida terminal foi prolongado em pacientes com disfunção hepática em comparação com o grupo controle de voluntários saudáveis (4,1-5 horas versus 2,8 horas). A redução da dose do medicamento em aproximadamente 50% é recomendada para pacientes com MF e PV que têm disfunção hepática (ver seção "Posologia e Administração").
Para pacientes com SMD e disfunção hepática não relacionada à SMD, a dose inicial de ruxolitinibe deve ser reduzida em 50%.
DADOS PRÉ-CLÍNICOS DE SEGURANÇA
O ruxolitinibe foi avaliado em estudos de farmacologia de segurança, toxicidade de doses repetidas, genotoxicidade e toxicidade reprodutiva, bem como em estudos de carcinogenicidade. Os órgãos-alvo relacionados à ação farmacológica do ruxolitinibe nos estudos de doses repetidas incluíram a medula óssea, o sangue periférico e os tecidos linfoides. Infecções comuns associadas à imunossupressão foram observadas em cães. A redução da pressão arterial e o aumento da frequência cardíaca foram observados em um estudo em cães com medições telemétricas; a redução do volume minuto respiratório foi observada em um estudo de funções do trato respiratório em ratos. Os limites de dose aceitáveis (com base nos níveis de Cmax do medicamento não ligado) nos estudos em cães e ratos foram, respectivamente, 15,7 e 10,4 vezes maiores do que a dose máxima recomendada para humanos de 25 mg duas vezes ao dia. Não se observaram efeitos colaterais na avaliação dos efeitos neuropsicofarmacológicos do ruxolitinibe. Nos estudos em animais, a administração de ruxolitinibe levou à redução do peso fetal e ao aumento da perda pós-implantacional. Não se encontraram evidências de efeitos teratogênicos nos estudos em ratos e coelhos. No entanto, os limites de exposição e a dose clínica mais alta foram baixos, então a interpretação dos resultados em termos de segurança para humanos é limitada. Não se observaram efeitos colaterais na fertilidade. Nos estudos de desenvolvimento pré e pós-natal em animais, foram observados um período de gestação ligeiramente prolongado, redução do número de implantações e nascimento de menos descendentes. Nos descendentes, foram observados níveis reduzidos de peso corporal inicial e um curto período de redução do ganho de peso corporal. Em ratas lactantes, o ruxolitinibe e/ou seus metabólitos foram eliminados no leite materno em concentrações 13 vezes maiores do que as concentrações no plasma sanguíneo das ratas lactantes. O ruxolitinibe não demonstrou propriedades mutagênicas ou clastogênicas. O ruxolitinibe não demonstrou propriedades carcinogênicas no modelo transgênico (camundongos da linha Tg.rasH2).
CARACTERÍSTICAS CLÍNICAS
INDICAÇÕES
Mielofibrose. Tratamento de doenças associadas à esplenomegalia ou sintomas de mielofibrose primária (também conhecida como mielofibrose idiopática crônica) em pacientes adultos, mielofibrose secundária à policitemia vera ou mielofibrose secundária à trombocitemia essencial.
Policitemia vera. Tratamento da policitemia vera em pacientes adultos com resistência ou intolerância à hidroxiureia.
Reação "transplante contra hospedeiro" (RTH). Tratamento de pacientes com idade igual ou superior a 12 anos com reação aguda de RTH ou reação crônica de RTH que têm resposta inadequada à terapia com corticosteroides ou outros medicamentos sistêmicos.
CONTRAINDICAÇÕES
Hipersensibilidade à substância ativa ou a qualquer um dos excipientes do medicamento. Período de gravidez e amamentação.
INTERAÇÕES COM OUTROS MEDICAMENTOS E OUTROS TIPOS DE INTERAÇÕES
Os estudos de interação foram realizados apenas com a participação de adultos.
O ruxolitinibe é eliminado do organismo por metabolismo, que é catalisado pelo CYP3A4 e CYP2C9. Portanto, a administração de medicamentos que inibem a ação dessas enzimas pode levar a um aumento da exposição ao ruxolitinibe.
INTERAÇÕES QUE REQUEREM REDUÇÃO DA DOSE DE RUXOLITINIBE
Inibidores do CYP3A4
Inibidores potentes do CYP3A4 (boceprevir, claritromicina, indinavir, itraconazol, cetoconazol, lopinavir/ritonavir, mibefradil, nefazodona, nelfinavir, posaconazol, saquinavir, telaprevir, telitromicina, voriconazol, etc.)
Em voluntários saudáveis, a administração concomitante de ruxolitinibe (dose única de 10 mg) com um inibidor potente do CYP3A4, cetoconazol, resultou em um aumento dos valores de Cmax e AUC do ruxolitinibe em 33% e 91%, respectivamente, em comparação com os valores de Cmax e AUC quando o ruxolitinibe foi administrado sozinho.
Quando o ruxolitinibe for administrado com inibidores potentes do CYP3A4, a dose padrão de ruxolitinibe deve ser reduzida em aproximadamente 50% e administrada duas vezes ao dia. Os pacientes devem ser cuidadosamente monitorados (por exemplo, duas vezes por semana) para o desenvolvimento de citopenia, e o ajuste da dose do medicamento deve ser feito com base na segurança e eficácia (ver seção "Posologia e Administração").
Inibidores duplos das enzimas CYP2C9 e CYP3A4
Em voluntários saudáveis, a administração concomitante de ruxolitinibe (dose única de 10 mg) com um inibidor duplo do CYP2C9 e CYP3A4, fluconazol, resultou em um aumento dos valores de Cmax e AUC do ruxolitinibe em 47% e 232%, respectivamente, em comparação com os valores de Cmax e AUC quando o ruxolitinibe foi administrado sozinho.
Deve-se considerar a redução da dose do medicamento em 50% quando administrado com medicamentos que sejam inibidores duplos das enzimas CYP2C9 e CYP3A4 (por exemplo, fluconazol). Deve-se evitar a administração de ruxolitinibe com fluconazol em doses superiores a 200 mg por dia.
OUTRAS INTERAÇÕES QUE AFETAM O RUXOLITINIBE
Inibidores leves ou moderados do CYP3A4 (ciprofloxacino, eritromicina, amprenavir, atazanavir, diltiazem, cimetidina, etc.)
Em voluntários saudáveis, a administração concomitante de ruxolitinibe (dose única de 10 mg) com eritromicina (500 mg duas vezes ao dia durante quatro dias) resultou em um aumento dos valores de Cmax e AUC do ruxolitinibe em 8% e 27%, respectivamente, em comparação com os valores de Cmax e AUC quando o ruxolitinibe foi administrado sozinho.
Quando o ruxolitinibe for administrado com inibidores leves ou moderados do CYP3A4 (por exemplo, eritromicina), não é recomendada a correção da dose do ruxolitinibe. No entanto, no início do tratamento com ruxolitinibe junto com um inibidor moderado da enzima CYP3A4, os pacientes devem ser cuidadosamente monitorados para a ocorrência de citopenia.
EFETOS DO RUXOLITINIBE SOBRE OUTROS MEDICAMENTOS
Substâncias transportadas pelo transporte de glicoproteína P ou outros transportadores
O ruxolitinibe pode inibir a atividade da glicoproteína P e da proteína de resistência ao câncer de mama (BCRP) no intestino. Isso pode levar a um aumento da exposição sistêmica dos substratos desses transportadores, como o etexilato de dabigatrana, ciclosporina, rosuvastatina e, possivelmente, digoxina. É recomendada a monitorização terapêutica ou clínica dos substratos envolvidos.
É possível que a inibição esperada da glicoproteína P e da proteína de resistência ao câncer de mama (BCRP) no intestino seja minimizada se o intervalo de tempo entre a administração dos medicamentos for o mais longo possível.
Estudos com voluntários saudáveis demonstraram que o ruxolitinibe não inibiu o metabolismo do midazolam administrado por via oral, que é um substrato do CYP3A4. Portanto, não se espera um aumento da exposição aos substratos do CYP3A4 quando administrados concomitantemente com o ruxolitinibe. Em outro estudo com voluntários saudáveis, verificou-se que o ruxolitinibe não afeta a farmacocinética dos anticoncepcionais orais que contêm etinilestradiol e levonorgestrel. Portanto, não se espera que o ruxolitinibe afete a eficácia dos anticoncepcionais orais quando administrados concomitantemente.
CARACTERÍSTICAS ESPECIAIS
MIELOSUPRESSÃO
O tratamento com o medicamento JAKAVI pode levar à ocorrência de reações adversas hematológicas, incluindo trombocitopenia, anemia e neutropenia. Antes de iniciar o tratamento com o medicamento JAKAVI, é necessário realizar um hemograma completo, incluindo contagem diferencial de leucócitos.
O tratamento deve ser interrompido em pacientes com contagem de plaquetas inferior a 50.000/mm3 (50 x 10^9/L) ou contagem absoluta de neutrófilos inferior a 500/mm3 (0,5 x 10^9/L) (ver seção "Posologia e Administração").
Foi determinado que os pacientes com MF e baixo nível de plaquetas (< 200.000/mm3 (< 200 x 10^9/L)) no início do tratamento são mais propensos a desenvolver trombocitopenia durante o tratamento.
A trombocitopenia, em geral, é reversível e, como regra, é corrigida pela redução da dose do medicamento ou interrupção temporária do tratamento com o medicamento JAKAVI (ver seções "Posologia e Administração" e "Reações Adversas"). No entanto, pode ser necessário realizar transfusões de plaquetas com base em indicações clínicas.
Pacientes com anemia podem precisar de transfusões de sangue. Para pacientes que desenvolvem anemia, pode ser necessária a alteração da dose do medicamento ou interrupção do tratamento.
Pacientes com nível de hemoglobina inferior a 10,0 g/dL (100 g/L) no início do tratamento têm um risco mais alto de redução do nível de hemoglobina para abaixo de 8 g/dL (80 g/L) durante o tratamento em comparação com pacientes com níveis mais altos de hemoglobina no início do tratamento (79,3% em comparação com 30,1%). Para pacientes com nível de hemoglobina inferior a 10,0 g/dL (100 g/L) no início do tratamento, é recomendada uma monitorização mais frequente dos parâmetros hematológicos e sintomas clínicos de reações adversas relacionadas ao medicamento JAKAVI.
A neutropenia (contagem absoluta de neutrófilos < 500 (< 0,5 x 10^9/L)), em geral, é reversível e, como regra, é corrigida pela interrupção temporária do tratamento com o medicamento JAKAVI (ver seções "Posologia e Administração" e "Reações Adversas"). Com base em indicações clínicas, é necessário realizar hemogramas completos, e as doses do medicamento devem ser ajustadas conforme necessário (ver seções "Posologia e Administração" e "Reações Adversas").
INFECÇÕES
Em pacientes que receberam o medicamento JAKAVI, foram observadas infecções graves, bacterianas, micobacterianas, fúngicas, virais e outras infecções oportunistas. O estado dos pacientes deve ser avaliado em relação ao risco de desenvolver infecções graves. Os médicos devem monitorar cuidadosamente os pacientes que recebem JAKAVI em relação a sintomas de infecções e iniciar tratamento apropriado rapidamente, se necessário. O tratamento com o medicamento JAKAVI não deve ser iniciado até que as infecções graves ativas sejam resolvidas.
Foi relatado tuberculose em pacientes que receberam JAKAVI. De acordo com as recomendações locais, antes de iniciar o tratamento, os pacientes devem ser examinados para detectar tuberculose ativa e latente ("oculta"), o que pode incluir estudo da história médica, determinação da possibilidade de contato prévio com pacientes com tuberculose e/ou realização de triagem apropriada, como radiografia de tórax, testes tuberculinizados e/ou análise de liberação de interferona gama, dependendo das circunstâncias. Existe o risco de resultados falsos negativos do teste tuberculinizado cutâneo, especialmente em pacientes com doenças graves ou imunidade comprometida.
Em pacientes com hepatite viral crônica B que receberam JAKAVI, foram observados aumento da carga viral (título de DNA do VHB) com aumento dos níveis de alanina aminotransferase e aspartato aminotransferase ou não. Antes de iniciar o tratamento com o medicamento JAKAVI, é recomendada a triagem para detectar VHB. Pacientes com infecção crônica por VHB devem receber tratamento e monitorização de acordo com as diretrizes clínicas.
HERPES ZÓSTER
Os médicos devem informar os pacientes sobre a ocorrência de sintomas precoces de herpes zóster, informando-os de que o tratamento deve ser iniciado o mais rápido possível.
LEUCOENCEFALOPATIA MULTIFOCAL PROGRESSIVA
Foi relatada leucoencefalopatia multifocal progressiva (LMP) em pacientes que receberam JAKAVI. Os médicos devem estar particularmente atentos a sintomas que possam indicar LMP, que os pacientes podem não notar (por exemplo, sintomas cognitivos, neurológicos ou psiquiátricos). Os pacientes devem ser monitorados em relação ao surgimento de novos ou piora dos sintomas existentes, e, se esses sintomas ocorrerem, deve-se considerar a necessidade de consultar um neurologista e realizar exames diagnósticos apropriados para detectar LMP. Se LMP for suspeitada, o tratamento com o medicamento deve ser interrompido até que a suspeita de LMP seja descartada.
DISFUNÇÃO/ALTERAÇÃO DO NÍVEL DE LÍPIDOS
O tratamento com o medicamento JAKAVI foi associado a alterações nos níveis de lípidos, incluindo aumento do colesterol total, colesterol de lipoproteína de alta densidade (LDL), colesterol de lipoproteína de baixa densidade (LDL) e triglicerídeos. É recomendada a monitorização dos níveis de lípidos e o tratamento da dislipidemia de acordo com as diretrizes clínicas.
REAÇÕES ADVERSAS CARDÍACAS PRINCIPAIS (MACE)
Em um grande estudo randomizado de tofacitinibe (outro inibidor de JAK) com controle ativo em pacientes com artrite reumatoide e pelo menos um fator adicional de risco de doenças cardiovasculares, foi observada uma frequência mais alta de MACE, definidos como morte cardiovascular, infarto do miocárdio não fatal e acidente vascular cerebral não fatal, com o uso de tofacitinibe em comparação com o uso de inibidores do fator de necrose tumoral (FNT).
Foram relatados casos de MACE em pacientes que receberam o medicamento JAKAVI. Antes de iniciar ou continuar o tratamento com o medicamento JAKAVI, é necessário avaliar cuidadosamente os benefícios e riscos para cada paciente individual, especialmente para pacientes com idade igual ou superior a 65 anos, pacientes que fumam ou têm história de fumo prolongado e pacientes com doenças cardiovasculares ateroscleróticas na história ou outros fatores de risco de doenças cardiovasculares na história.
TROMBOSE
Em um grande estudo randomizado de tofacitinibe (outro inibidor de JAK) com controle ativo em pacientes com artrite reumatoide e pelo menos um fator adicional de risco de doenças cardiovasculares, foi observada uma frequência mais alta de tromboembolia venosa (TEV), incluindo trombose venosa profunda (TVP) e embolia pulmonar (EP), com o uso de tofacitinibe em comparação com o uso de inibidores do FNT.
Foram relatados casos de trombose em pacientes que receberam o medicamento JAKAVI. Em estudos clínicos com pacientes com mielofibrose e policitemia vera, a frequência de tromboembolia foi comparável entre o grupo que recebeu o medicamento JAKAVI e o grupo de controle.
Antes de iniciar ou continuar o tratamento com o medicamento JAKAVI, é necessário avaliar cuidadosamente os benefícios e riscos para cada paciente individual, especialmente para pacientes com fatores de risco de doenças cardiovasculares (também ver "Reações Adversas Cardíacas Principais (MACE)" acima).
Pacientes com sintomas de trombose devem ser examinados imediatamente e receber tratamento apropriado.
NEOPLASIAS SECUNDÁRIAS, DETECTADAS PELA PRIMEIRA VEZ
Em um grande estudo randomizado de tofacitinibe (outro inibidor de JAK) com controle ativo em pacientes com artrite reumatoide e pelo menos um fator adicional de risco de doenças cardiovasculares, foi observada uma frequência mais alta de neoplasias, especialmente câncer de pulmão, linfoma e câncer de pele não melanoma (CPNM), com o uso de tofacitinibe em comparação com o uso de inibidores do FNT.
Foram relatados casos de linfoma e outras neoplasias em pacientes que receberam inibidores de JAK, incluindo o medicamento JAKAVI.
CPNM, incluindo carcinoma basocelular, carcinoma de células escamosas e carcinoma de células de Merkel, foi observado em pacientes que receberam ruxolitinibe. A maioria desses pacientes com MF e PV tinha história de tratamento prolongado com hidroxiureia e história de neoplasias prévias ou lesões pré-malignas na pele. É recomendada a realização de exames de pele periódicos em pacientes com risco aumentado de câncer de pele.
GRUPOS ESPECIAIS DE PACIENTES
PACIENTES COM DISFUNÇÃO RENAL
A dose inicial do medicamento JAKAVI deve ser reduzida para pacientes com disfunção renal grave. Para pacientes com MF e estágio terminal de doença renal em diálise, a dose inicial do medicamento deve ser determinada com base na contagem de plaquetas, enquanto a dose inicial recomendada para pacientes com PV é de 10 mg uma vez ao dia (ver seção "Posologia e Administração"). As doses subsequentes do medicamento (dose única de 20 mg ou duas doses de 10 mg, administradas com intervalo de 12 horas, separadamente, para pacientes com MF; dose única de 10 mg ou duas doses de 5 mg, administradas com intervalo de 12 horas, para pacientes com PV) devem ser administradas apenas nos dias de diálise, após a conclusão de cada sessão de diálise. Alterações adicionais na posologia do medicamento devem ser feitas com monitorização cuidadosa da segurança e eficácia do medicamento (ver seções "Posologia e Administração" e "Farmacocinética").
PACIENTES COM DISFUNÇÃO HEPÁTICA
A dose inicial do medicamento JAKAVI deve ser reduzida em aproximadamente 50% para pacientes com MF ou PV e disfunção hepática. Alterações subsequentes da dose do medicamento devem ser feitas com base na segurança e eficácia do medicamento. Para pacientes com RTH e disfunção hepática não relacionada à RTH, a dose inicial do medicamento JAKAVI deve ser reduzida em aproximadamente 50% (ver seções "Posologia e Administração" e "Farmacocinética").
INTERAÇÕES
Se o JAKAVI for administrado concomitantemente com inibidores potentes do CYP3A4 ou inibidores duplos das enzimas CYP2C9 e CYP3A4 (por exemplo, fluconazol), a dose padrão de JAKAVI deve ser reduzida em aproximadamente 50% e administrada duas vezes ao dia (informações sobre a frequência de monitorização estão nas seções "Posologia e Administração" e "Interações com Outros Medicamentos e Outros Tipos de Interações").
A administração concomitante de terapia citorredutora com o medicamento JAKAVI foi associada à ocorrência de citopenia tratável (ver seção "Posologia e Administração").
SÍNDROME DE ABSTINÊNCIA DO MEDICAMENTO
Sintomas de MF podem retornar dentro de aproximadamente uma semana após a interrupção temporária ou a suspensão do tratamento com o medicamento JAKAVI. Foram relatados casos de abstinência do medicamento JAKAVI entre pacientes que sofreram reações adversas graves, especialmente quando havia doença aguda concomitante. Até o momento, não foi estabelecido se a interrupção abrupta do tratamento com o medicamento JAKAVI contribui para o desenvolvimento desses fenômenos. Além dos casos em que a interrupção abrupta do tratamento é necessária, deve-se considerar a possibilidade de reduzir gradualmente a dose do JAKAVI, embora o benefício dessa redução gradual da dose não tenha sido comprovado.
EXCIPIENTES
O JAKAVI contém lactose. Pacientes com intolerância conhecida a certains açúcares devem consultar um médico antes de tomar este medicamento. Pacientes com doenças raras hereditárias, como intolerância à galactose, deficiência grave de lactase ou má absorção de glicose-galactose, não devem tomar este medicamento.
Este medicamento contém menos de 1 mmol de sódio (23 mg) por dose, portanto, é considerado como não contendo sódio.
USO DURANTE A GRAVIDEZ OU AMAMENTAÇÃO
GRAVIDEZ
Os dados sobre o uso do medicamento JAKAVI em mulheres grávidas são limitados. Estudos em animais mostraram que o ruxolitinibe é embriotóxico e fetotóxico. A teratogenicidade não foi observada em ratos ou coelhos. No entanto, os limites de exposição e a dose clínica mais alta foram baixos, então a interpretação dos resultados em termos de segurança para humanos é limitada. O risco potencial para humanos é desconhecido. Como medida de precaução, o uso do medicamento JAKAVI durante a gravidez é contraindicado (ver seção "Contraindicações").
MULHERES EM IDADE REPRODUTIVA/CONTRACEPÇÃO
Mulheres em idade reprodutiva devem usar métodos contraceptivos eficazes durante o tratamento com o medicamento JAKAVI. Em caso de gravidez durante o tratamento com o medicamento JAKAVI, a avaliação do risco/benefício deve ser feita individualmente, com consideração cuidadosa dos riscos potenciais para o feto.
AMAMENTAÇÃO
O medicamento JAKAVI não deve ser usado durante a amamentação, portanto, se o tratamento for iniciado, a amamentação deve ser interrompida. É desconhecido se o ruxolitinibe e/ou seus metabólitos são excretados no leite materno humano. O risco para a criança amamentada não pode ser excluído. Os dados farmacodinâmicos/toxicológicos disponíveis de estudos em animais mostram que o ruxolitinibe e seus metabólitos são excretados no leite materno de animais.
FERTILIDADE
Os dados sobre o efeito do ruxolitinibe na fertilidade humana são limitados. O efeito do medicamento na fertilidade não foi observado em estudos em animais.
CAPACIDADE DE INFLUENCIAR A VELOCIDADE DE REAÇÃO AO DIRIGIR VEÍCULOS OU OPERAR MÁQUINAS
O JAKAVI não tem ou tem um efeito sedativo mínimo. No entanto, pacientes que experimentam tontura após a administração do JAKAVI devem abster-se de dirigir veículos ou operar máquinas.
POSologia E ADMINISTRAÇÃO
O medicamento JAKAVI deve ser administrado por via oral, independentemente da ingestão de alimentos.
Se uma dose for perdida, o paciente não deve tomar uma dose adicional, e a próxima dose deve ser administrada no horário usual.
O tratamento com o medicamento JAKAVI deve ser iniciado por um médico com experiência no uso de medicamentos antineoplásicos.
Antes de iniciar o tratamento com o medicamento JAKAVI, é necessário realizar um hemograma completo, incluindo contagem diferencial de leucócitos.
Os parâmetros do hemograma, incluindo a contagem diferencial de leucócitos, devem ser monitorados a cada 2-4 semanas, e, em seguida, conforme necessário clínico, até a estabilização da dose do medicamento JAKAVI (ver seção "Características Especiais").
DOSE
Dose inicial
A dose inicial recomendada do medicamento JAKAVI para a mielofibrose (MF) é determinada com base na contagem de plaquetas (ver tabela "Doses iniciais para mielofibrose").
| Contagem de plaquetas | Dose inicial |
| Above 200.000/mm3 (> 200 x 10^9/L) | 20 mg por via oral duas vezes ao dia |
| De 100.000 a 200.000/mm3 (100 x 10^9/L - 200 x 10^9/L) | 15 mg por via oral duas vezes ao dia |
| De 75.000 a menos de 100.000/mm3 (75 x 10^9/L - < 100 x 10^9/L) | 10 mg por via oral duas vezes ao dia |
| De 50.000 a menos de 75.000/mm3 (50 x 10^9/L - < 75 x 10^9/L) | 5 mg por via oral duas vezes ao dia |
A dose inicial recomendada do medicamento JAKAVI para a policitemia vera (PV) é de 10 mg por via oral duas vezes ao dia.
A dose inicial recomendada do medicamento JAKAVI para a reação "transplante contra hospedeiro" (RTH) aguda e crônica é de 10 mg por via oral duas vezes ao dia. O JAKAVI pode ser administrado como adjuvante à terapia de manutenção com corticosteroides e/ou inibidores da calcineurina (ICN).
AJUSTE DA DOSE
As doses podem ser tituladas com base na segurança e eficácia do medicamento.
Mielofibrose e policitemia vera
Se a eficácia do medicamento for considerada insuficiente, mas os parâmetros hematológicos estiverem em níveis adequados, as doses do medicamento podem ser aumentadas de 5 mg duas vezes ao dia para uma dose máxima de 25 mg duas vezes ao dia.
A dose inicial do medicamento não deve ser aumentada durante as primeiras quatro semanas de tratamento, e, em seguida, o aumento deve ser feito em intervalos de não menos de 2 semanas.
O tratamento deve ser interrompido se a contagem de plaquetas for inferior a 50.000/mm3 (50 x 10^9/L) ou a contagem absoluta de neutrófilos for inferior a 500/mm3 (0,5 x 10^9/L). No caso de PV, o tratamento também deve ser interrompido se o nível de hemoglobina for inferior a 8 g/dL (80 g/L). Após a normalização dos parâmetros hematológicos acima desses níveis, a administração do medicamento pode ser retomada com uma dose de 5 mg duas vezes ao dia, com aumento gradual da dose com base no monitoramento cuidadoso dos parâmetros do hemograma, incluindo a contagem diferencial de leucócitos.
Deve-se reduzir a dose se, durante o tratamento, a contagem de plaquetas diminuir, como indicado na tabela "Recomendações de dosagem para pacientes com MF e trombocitopenia", com o objetivo de evitar a interrupção do tratamento devido à trombocitopenia.
| Contagem de plaquetas | Dose na redução da plaqueta |
| De 100.000 a < 125.000/mm3 (100 x 10^9/L - 125 x 10^9/L) | 20 mg duas vezes ao dia |
| De 75.000 a < 100.000/mm3 (75 x 10^9/L - 100 x 10^9/L) | 15 mg duas vezes ao dia |
| De 50.000 a < 75.000/mm3 (50 x 10^9/L - 75 x 10^9/L) | 10 mg duas vezes ao dia |
| Menos de 50.000/mm3 (< 50 x 10^9/L) | Tratamento deve ser interrompido |
Reação "transplante contra hospedeiro"
A redução da dose e a interrupção do tratamento devem ser consideradas em relação a pacientes com RTH e trombocitopenia, neutropenia ou aumento do nível de bilirrubina total após terapia de manutenção padrão, incluindo fatores de crescimento, agentes anti-infecciosos e transfusões de sangue. É recomendada a redução da dose em um nível (de 10 mg duas vezes ao dia para 5 mg duas vezes ao dia ou de 5 mg duas vezes ao dia para 5 mg uma vez ao dia). Pacientes que não toleram o JAKAVI na dose de 5 mg uma vez ao dia devem ter o tratamento interrompido. As recomendações detalhadas para a dosagem estão na tabela "Recomendações de dosagem durante a terapia com ruxolitinibe em pacientes com RTH e trombocitopenia, neutropenia ou aumento do nível de bilirrubina total".
| Parâmetro laboratorial | Recomendações de dosagem |
| Contagem de plaquetas < 20.000/mm3 (< 20 x 10^9/L) | Reduzir a dose do JAKAVI em um nível. Se a contagem de plaquetas permanecer ≥ 20.000/mm3 (≥ 20 x 10^9/L) durante 7 dias, a dose pode ser aumentada para o nível inicial; caso contrário, a dose reduzida deve ser mantida. |
| Contagem de plaquetas < 15.000/mm3 (< 15 x 10^9/L) | Interromper a administração do medicamento JAKAVI até que a contagem de plaquetas atinja ≥ 20.000/mm3 (≥ 20 x 10^9/L), e, em seguida, retomar a administração do medicamento com a dose reduzida em um nível. |
| Contagem absoluta de neutrófilos (CAN) de ≥ 500/mm3 a < 750/mm3 (0,5 x 10^9/L - 0,75 x 10^9/L) | Reduzir a dose do JAKAVI em um nível. Em seguida, retomar a administração do medicamento com a dose inicial se a CAN > 1.000/mm3 (> 1 x 10^9/L). |
| Contagem absoluta de neutrófilos < 500/mm3 (< 0,5 x 10^9/L) | Interromper a administração do JAKAVI até que a CAN > 500/mm3 (> 0,5 x 10^9/L), e, em seguida, retomar a administração do medicamento com a dose reduzida em um nível. Se a CAN > 1.000/mm3 (> 1 x 10^9/L), retomar a administração do medicamento com a dose inicial. |
AJUSTE DA DOSE DO MEDICAMENTO NA ADMINISTRAÇÃO CONCOMITANTE COM INIBIDORES POTENTES DO CYP3A4 OU INIBIDORES DUPLA DAS ENZIMAS CYP2C9/3A4
Quando o ruxolitinibe for administrado com inibidores potentes do CYP3A4 ou inibidores duplos das enzimas CYP2C9 e CYP3A4 (por exemplo, fluconazol), a dose padrão do ruxolitinibe deve ser reduzida em aproximadamente 50% e administrada duas vezes ao dia (ver seção "Interações com Outros Medicamentos e Outros Tipos de Interações").
Deve-se evitar a administração do ruxolitinibe com fluconazol em doses superiores a 200 mg por dia.
Durante a administração concomitante do medicamento com inibidores potentes do CYP3A4 ou inibidores duplos das enzimas CYP2C9 e CYP3A4, é recomendada a realização de monitoramento mais frequente (por exemplo, duas vezes por semana) dos parâmetros hematológicos e sintomas clínicos de reações adversas relacionadas ao JAKAVI.
GRUPOS ESPECIAIS DE PACIENTES
Disfunção renal
Pacientes com disfunção renal leve ou moderada não necessitam de ajuste da dose do medicamento.
Para pacientes com disfunção renal grave (depuração de creatinina < 30 mL/min), a dose inicial recomendada com base na contagem de plaquetas para a MF deve ser reduzida em aproximadamente 50% e administrada duas vezes ao dia. A dose inicial recomendada para pacientes com PV é de 5 mg duas vezes ao dia. Com base na segurança e eficácia, os pacientes devem ser monitorados cuidadosamente durante o tratamento com o medicamento JAKAVI.
Existem apenas dados limitados sobre a dosagem para pacientes com estágio terminal de doença renal em diálise.
Disfunção hepática
Para pacientes com MF e qualquer disfunção hepática, a dose inicial recomendada com base na contagem de plaquetas deve ser reduzida em aproximadamente 50% e administrada duas vezes ao dia. As doses subsequentes do medicamento devem ser ajustadas com base na segurança e eficácia. A dose inicial recomendada para pacientes com PV é de 5 mg duas vezes ao dia.
Pacientes com disfunção hepática leve, moderada ou grave não relacionada à RTH devem ter a dose inicial do ruxolitinibe reduzida em 50% (ver seção "Farmacocinética").
Pacientes com disfunção hepática relacionada à RTH e aumento do nível de bilirrubina total > 3 x LSN devem realizar análises de sangue com mais frequência para detectar toxicidade. É recomendada a redução da dose em um nível.
PACIENTES IDOSOS (≥ 65 ANOS)
Pacientes idosos não necessitam de ajuste adicional da dose do medicamento.
INTERRUPÇÃO DO TRATAMENTO
O tratamento de MF e PV com o medicamento JAKAVI pode ser prolongado desde que a relação benefício/risco permaneça favorável. No entanto, o tratamento deve ser interrompido após 6 meses se não houver redução do tamanho do baço ou alívio dos sintomas após o início da terapia.
Pacientes que demonstraram algum grau de melhora clínica devem ter o tratamento com ruxolitinibe interrompido se houver aumento adicional do tamanho do baço em 40% em relação ao tamanho basal (o que é aproximadamente equivalente a um aumento de 25% no volume do baço) e não houver alívio adicional dos sintomas relacionados à doença.
Para a RTH, deve-se considerar a possibilidade de reduzir gradualmente a dose do medicamento JAKAVI em pacientes que respondem ao tratamento e após a interrupção da administração de corticosteroides. É recomendada a redução da dose do medicamento JAKAVI em 50% a cada 2 meses. Deve-se considerar a possibilidade de aumentar a dose novamente se os sinais ou sintomas de RTH reaparecerem durante ou após a redução da dose do medicamento JAKAVI.
CRIANÇAS
A segurança e eficácia do medicamento JAKAVI em crianças (menores de 18 anos) com MF e PV não foram estabelecidas. Os dados são limitados (ver seção "Farmacodinâmica").
A segurança e eficácia do JAKAVI em crianças com idade igual ou superior a 12 anos com RTH são suportadas por dados de estudos randomizados de fase 3 REACH2 e REACH3. A dose do medicamento JAKAVI para crianças com RTH é a mesma que para adultos. A segurança e eficácia do medicamento JAKAVI em crianças menores de 12 anos não foram estabelecidas.
SUPERDOSE
O antídoto para a superdose do medicamento JAKAVI é desconhecido. A administração de doses únicas do medicamento de até 200 mg foi bem tolerada. A administração de doses mais altas do que as doses repetidas recomendadas foi associada a níveis aumentados de mielossupressão, incluindo leucopenia, anemia e trombocitopenia. Nesses casos, deve-se aplicar terapia de suporte apropriada. Não se espera que a realização de diálise acelere a eliminação do ruxolitinibe do organismo.
Reações Adversas
Resumo do Perfil de Segurança
Mielofibrose
As reações adversas mais frequentemente relatadas foram trombocitopenia e anemia.
As reações adversas hematológicas (de qualquer grau, de acordo com os Critérios Comuns de Terminologia para Eventos Adversos [CTCAE]) incluíram anemia (83,8%), trombocitopenia (80,5%) e neutropenia (20,8%).
Anemia, trombocitopenia e neutropenia são efeitos dose-dependentes.
As três reações adversas não hematológicas mais frequentes foram equimoses (33,3%), outras hemorragias (incluindo epistaxe, hemorragias pós-procedimento e hematúria) (24,3%) e tontura (21,9%).
As três anomalias laboratoriais não hematológicas mais frequentes foram níveis elevados de alanina aminotransferase (40,7%), níveis elevados de aspartato aminotransferase (31,5%) e hipertrigliceridemia (25,2%). Nos estudos clínicos de fase 3 em pacientes com MF, não houve hipertrigliceridemia ou elevação do nível de aspartato aminotransferase de grau 3 ou 4, de acordo com a CTCAE, e não houve elevação do nível de alanina aminotransferase ou hipercolesterolemia de grau 4, de acordo com a CTCAE.
A interrupção do tratamento devido a reações adversas, independentemente da relação de causa e efeito, ocorreu em 30,0% dos pacientes.
Policiemia Vera
As reações adversas mais frequentemente relatadas foram anemia e níveis elevados de alanina aminotransferase.
As reações adversas hematológicas (de qualquer grau, de acordo com a CTCAE) incluíram anemia (61,8%), trombocitopenia (25,0%) e neutropenia (5,3%). Anemia e trombocitopenia de grau 3 ou 4, de acordo com a CTCAE, ocorreram em 2,9% ou 2,6% dos pacientes, respectivamente.
As três reações adversas não hematológicas mais frequentes foram aumento de peso (20,3%), tontura (19,4%) e cefaleia (17,9%).
As três anomalias laboratoriais não hematológicas mais frequentes (de qualquer grau, de acordo com a CTCAE) identificadas como reações adversas foram níveis elevados de alanina aminotransferase (45,3%), níveis elevados de aspartato aminotransferase (42,6%) e hipercolesterolemia (34,7%). Não houve níveis elevados de alanina aminotransferase ou hipercolesterolemia de grau 4, de acordo com a CTCAE, e houve um caso de nível elevado de aspartato aminotransferase de grau 4, de acordo com a CTCAE.
A interrupção do tratamento devido a reações adversas, independentemente da relação de causa e efeito, ocorreu em 19,4% dos pacientes.
Trombocitopenia Refratária
As reações adversas mais frequentemente relatadas foram trombocitopenia, anemia e neutropenia.
As anomalias laboratoriais hematológicas identificadas como reações adversas incluíram trombocitopenia (85,2%), anemia (75,0%) e neutropenia (65,1%). Anemia de grau 3 foi relatada em 47,7% dos pacientes (grau 4 não é previsto de acordo com a CTCAE versão 4.03). Trombocitopenia de graus 3 e 4 foi observada em 31,3% e 47,7% dos pacientes, respectivamente.
As três reações adversas não hematológicas mais frequentes incluíram infecção por citomegalovirus (32,3%), sepse (25,4%) e infecções do trato urinário (17,9%).
As três anomalias laboratoriais não hematológicas mais frequentes identificadas como reações adversas foram níveis elevados de alanina aminotransferase (54,9%), níveis elevados de aspartato aminotransferase (52,3%) e hipercolesterolemia (49,2%). A maioria delas foi de graus 1 e 2 de severidade.
A interrupção do tratamento devido a eventos adversos, independentemente da relação de causa e efeito, ocorreu em 29,4% dos pacientes.
Trombocitopenia Crônica
As reações adversas mais frequentemente relatadas foram anemia, hipercolesterolemia e níveis elevados de aspartato aminotransferase.
As anomalias laboratoriais hematológicas identificadas como reações adversas incluíram anemia (68,6%), trombocitopenia (34,4%) e neutropenia (36,2%). Anemia de grau 3 foi relatada em 14,8% dos pacientes (grau 4 não é previsto de acordo com a CTCAE versão 4.03). Neutropenia de graus 3 e 4 foi observada em 9,5% e 6,7% dos pacientes, respectivamente.
As três reações adversas não hematológicas mais frequentes incluíram hipertensão arterial (15,0%), cefaleia (10,2%) e infecções do trato urinário (9,3%).
As três anomalias laboratoriais não hematológicas mais frequentes identificadas como reações adversas foram hipercolesterolemia (52,3%), níveis elevados de aspartato aminotransferase (52,2%) e níveis elevados de alanina aminotransferase (43,1%). A maioria delas foi de graus 1 e 2 de severidade.
A interrupção do tratamento devido a eventos adversos, independentemente da relação de causa e efeito, ocorreu em 18,1% dos pacientes.
Resumo das Reações Adversas durante os Estudos Clínicos
A segurança em pacientes com MF foi avaliada com base nos dados de longo prazo de dois estudos de fase 3 (COMFORT I e COMFORT II), incluindo dados de pacientes que foram inicialmente randomizados para o grupo de ruxolitinibe (n = 301) e pacientes que receberam ruxolitinibe após a transição do grupo de controle (n = 156). A mediana de exposição, na qual se baseiam as categorias de frequência de reações adversas ao medicamento, em pacientes com MF é de 30,5 meses (variação de 0,3 a 68,1 meses).
A segurança em pacientes com PV foi avaliada com base nos dados de longo prazo de dois estudos de fase 3 (RESPONSE, RESPONSE 2), incluindo dados de pacientes que foram inicialmente randomizados para o grupo de ruxolitinibe (n = 184) e pacientes que receberam ruxolitinibe após a transição do grupo de controle (n = 156). A mediana de exposição, na qual se baseiam as categorias de frequência de reações adversas ao medicamento, em pacientes com PV é de 41,7 meses (variação de 0,03 a 59,7 meses).
A segurança do medicamento Jakavi em pacientes com TRT aguda foi avaliada no estudo de fase 3 REACH2, incluindo dados de pacientes que foram inicialmente randomizados para o grupo de medicamento Jakavi (n = 152) e pacientes que receberam medicamento Jakavi após a transição do grupo de melhor tratamento disponível (n = 49). A mediana de exposição, na qual se baseiam as categorias de frequência de reações adversas ao medicamento, foi de 8,9 semanas (variação de 0,3 a 66,1 semanas).
A segurança do medicamento Jakavi em pacientes com TRT crônica foi avaliada no estudo de fase 3 REACH3, incluindo dados de pacientes que foram inicialmente randomizados para o grupo de medicamento Jakavi (n = 165) e pacientes que receberam medicamento Jakavi após a transição do grupo de melhor tratamento disponível (n = 61). A mediana de exposição, na qual se baseiam as categorias de frequência de reações adversas ao medicamento, foi de 41,4 semanas (variação de 0,7 a 127,3 semanas).
No programa de estudos clínicos, a gravidade das reações adversas foi avaliada com base na CTCAE, que define o grau 1 como leve, grau 2 como moderado, grau 3 como grave, grau 4 como potencialmente fatal ou incapacitante e grau 5 como fatal.
Frequência de Reações Adversas em Estudos de Fase 3 com Pacientes com MF e PV
| Reações Adversas | Frequência em Pacientes com MF | Frequência em Pacientes com PV |
| Infecções e Infestações | ||
| Infecções do Trato Urinário | Muito Comum | Muito Comum |
| Herpes Zóster | Muito Comum | Muito Comum |
| Pneumonia | Muito Comum | Comum |
| Sepse | Comum | Pouco Comum |
| Tuberculose | Pouco Comum | Desconhecido |
| Reativação do Vírus da Hepatite B | Desconhecido | Pouco Comum |
| Distúrbios do Sangue e do Sistema Linfático | ||
| Anemia | ||
| Grau 4, de acordo com a CTCAE | Muito Comum | Pouco Comum |
| Grau 3, de acordo com a CTCAE | Muito Comum | Comum |
| Qualquer Grau, de acordo com a CTCAE | Muito Comum | Muito Comum |
| Trombocitopenia | ||
| Grau 4, de acordo com a CTCAE | Comum | Pouco Comum |
| Grau 3, de acordo com a CTCAE | Muito Comum | Comum |
| Qualquer Grau, de acordo com a CTCAE | Muito Comum | Muito Comum |
| Neutropenia | ||
| Grau 4, de acordo com a CTCAE | Comum | Pouco Comum |
| Grau 3, de acordo com a CTCAE | Comum | Pouco Comum |
| Qualquer Grau, de acordo com a CTCAE | Muito Comum | Comum |
| Pancitopenia | Comum | Comum |
| Hemorragias (Qualquer Hemorragia, Incluindo Hemorragias Intracranianas, Hemorragias Gastrointestinais, Equimoses e Outras Hemorragias) | Muito Comum | Muito Comum |
| Equimoses | Muito Comum | Muito Comum |
| Hemorragias Gastrointestinais | Muito Comum | Comum |
| Hemorragias Intracranianas | Comum | Pouco Comum |
| Outras Hemorragias (Incluindo Epistaxe, Hemorragias Pós-Procedimento e Hematúria) | Muito Comum | Muito Comum |
| Distúrbios Metabólicos e Nutricionais | ||
| Hipercolesterolemia | Muito Comum | Muito Comum |
| Hipertrigliceridemia | Muito Comum | Muito Comum |
| Aumento de Peso | Muito Comum | Muito Comum |
| Distúrbios do Sistema Nervoso | ||
| Tontura | Muito Comum | Muito Comum |
| Cefaleia | Muito Comum | Muito Comum |
| Distúrbios Gastrointestinais | ||
| Aumento da Lipase | Muito Comum | Muito Comum |
| Constipação | Muito Comum | Muito Comum |
| Flatulência | Comum | Comum |
| Distúrbios Hepáticos e da Vesícula Biliar | ||
| Aumento da Alanina Aminotransferase | Muito Comum | Muito Comum |
| Aumento da Aspartato Aminotransferase | Muito Comum | Muito Comum |
| Distúrbios Cardiovasculares | ||
| Hipertensão Arterial | Muito Comum | Muito Comum |
Frequência de Reações Adversas em Estudos de Fase 3 com Pacientes com TRT
| Reações Adversas | TRT Aguda (REACH2) | TRT Crônica (REACH3) |
| Infecções e Infestações | Comum | Comum |
| Infecção por Citomegalovirus | Muito Comum | Comum |
| Sepse | Muito Comum | - |
| Infecções do Trato Urinário | Muito Comum | Comum |
| Infecção por Vírus BK | - | Comum |
| Distúrbios do Sangue e do Sistema Linfático | ||
| Trombocitopenia | Muito Comum | Muito Comum |
| Anemia | Muito Comum | Muito Comum |
| Neutropenia | Muito Comum | Muito Comum |
| Pancitopenia | Muito Comum | - |
| Distúrbios Metabólicos e Nutricionais | ||
| Hipercolesterolemia | Muito Comum | Muito Comum |
| Aumento de Peso | - | Comum |
| Distúrbios do Sistema Nervoso | ||
| Cefaleia | Comum | Muito Comum |
| Distúrbios Cardiovasculares | ||
| Hipertensão Arterial | Muito Comum | Muito Comum |
| Distúrbios Gastrointestinais | ||
| Aumento da Lipase | - | Muito Comum |
| Aumento da Amilase | - | Muito Comum |
| Náusea | Muito Comum | - |
| Constipação | - | Comum |
| Distúrbios Hepáticos e da Vesícula Biliar | ||
| Aumento da Alanina Aminotransferase | Muito Comum | Muito Comum |
| Aumento da Aspartato Aminotransferase | Muito Comum | Muito Comum |
| Distúrbios Musculoesqueléticos e do Tecido Conjuntivo | ||
| Aumento da Creatina Quinase | - | Muito Comum |
| Distúrbios Renais e do Trato Urinário | ||
| Aumento da Creatinina | - | Muito Comum |
| 1 A frequência é definida com base em novas ou exacerbadas anomalias laboratoriais em relação ao valor basal. | ||
| 2 A pancitopenia é definida como hemoglobina < 100 g/L, contagem de plaquetas < 100 x 10^9/L e contagem de neutrófilos < 1,5 x 10^9/L (ou leucopenia de grau 2, se a contagem de neutrófilos não for estabelecida) simultaneamente em uma única avaliação laboratorial. | ||
| 3 CTCAE versão 4.03. | ||
| 4 A sepse de grau ≥ 3 inclui 20 (10%) casos de doença de grau 5. | ||
| 5 Dados não disponíveis: não foi relatado nenhum caso. | ||
Descrição de Reações Adversas Específicas
Anemia
Nos estudos clínicos de fase 3 em pacientes com MF, a mediana do tempo até o primeiro evento de anemia de grau 2 ou superior, de acordo com a CTCAE, foi de 1,5 meses. Um paciente (0,3%) interrompeu o tratamento devido ao desenvolvimento de anemia.
Nos pacientes que receberam ruxolitinibe, a média da redução máxima do nível de hemoglobina em relação ao valor basal foi de 10 g/L após 8-12 semanas de tratamento, após o que o nível de hemoglobina se recuperou gradualmente para um novo nível de equilíbrio, que foi aproximadamente 5 g/L menor que o valor basal. Essa tendência foi observada em pacientes independentemente de terem recebido transfusão de sangue durante o tratamento.
No estudo randomizado e controlado com placebo COMFORT I, 60,6% dos pacientes com MF que receberam ruxolitinibe e 37,7% dos pacientes com MF que receberam placebo necessitaram de transfusão de massa eritrocitária durante o período de tratamento randomizado. A frequência de transfusão de massa eritrocitária foi de 53,4% no grupo de ruxolitinibe e 41,1% no grupo de tratamento ótimo disponível no estudo COMFORT II.
No período randomizado dos estudos de fase 3, a anemia foi relatada com menos frequência em pacientes com PV (40,8%) em comparação com pacientes com MF (82,4%). A frequência de eventos de grau 3 e 4, de acordo com a CTCAE, foi de 2,7% na população de pacientes com PV, enquanto na população de pacientes com MF foi de 42,56%.
Durante os estudos de fase 3 com pacientes com TRT aguda e crônica, a anemia de grau 3, de acordo com a CTCAE, foi relatada em 47,7% e 14,8% dos pacientes, respectivamente.
Trombocitopenia
Nos estudos clínicos de fase 3 em pacientes com MF, a mediana do tempo até o primeiro evento de trombocitopenia de grau 3 ou 4 foi de aproximadamente 8 semanas. A trombocitopenia foi geralmente reversível após a redução da dose ou interrupção do tratamento. A mediana do tempo até a recuperação da contagem de plaquetas acima de 50.000/μL foi de 14 dias. Durante o período randomizado, a transfusão de massa plaquetária foi necessária em 4,7% dos pacientes que receberam ruxolitinibe e 4,0% dos pacientes que receberam esquemas de tratamento de controle. A interrupção do tratamento devido à trombocitopenia ocorreu em 0,7% dos pacientes que receberam ruxolitinibe e 0,9% dos pacientes que receberam esquemas de tratamento de controle. Em pacientes com contagem de plaquetas basal entre 100.000-200.000/μL, a frequência de trombocitopenia de grau 3 ou 4 foi maior em comparação com pacientes com contagem de plaquetas basal > 200.000/μL (64,2% versus 38,5%).
No período randomizado dos estudos de fase 3, a trombocitopenia ocorreu com menos frequência em pacientes com PV (16,8%) em comparação com pacientes com MF (69,8%). A frequência de eventos graves (grau 3 ou 4, de acordo com a CTCAE) de trombocitopenia em pacientes com PV (2,7%) foi menor do que em pacientes com MF (11,6%).
Nos estudos de fase 3 com pacientes com TRT aguda, a trombocitopenia de grau 3 e 4 ocorreu em 31,3% e 47,7% dos pacientes, respectivamente. No estudo de fase 3 com pacientes com TRT crônica, a frequência de desenvolvimento de trombocitopenia de grau 3 e 4 foi menor (5,9% e 10,7%, respectivamente) do que em pacientes com TRT aguda.
Neutropenia
Nos estudos clínicos de fase 3 em pacientes com MF, a mediana do tempo até o primeiro evento de neutropenia de grau 3 ou 4 foi de aproximadamente 12 semanas. Durante o período randomizado, a interrupção do tratamento ou redução da dose devido à neutropenia foi relatada em 1,0% dos pacientes, e a interrupção do tratamento devido à neutropenia ocorreu em 0,3% dos pacientes.
No período randomizado dos estudos de fase 3, a neutropenia foi relatada em 1,6% dos pacientes com PV que receberam ruxolitinibe, em comparação com 7% dos pacientes no grupo de controle. No grupo de ruxolitinibe, um paciente desenvolveu neutropenia de grau 4, de acordo com a CTCAE. Durante o acompanhamento de longo prazo nos estudos de fase 3, a neutropenia de grau 4, de acordo com a CTCAE, foi relatada em 2 pacientes.
Nos estudos de fase 3 com pacientes com TRT aguda, a neutropenia de grau 3 e 4 ocorreu em 17,9% e 20,6% dos pacientes, respectivamente. Nos estudos de fase 3 com pacientes com TRT crônica, a frequência de desenvolvimento de neutropenia de grau 3 e 4 foi menor (9,5% e 6,7%, respectivamente) do que em pacientes com TRT aguda.
Hemorragias
Nos estudos de fase 3, a frequência de eventos de hemorragia (incluindo hemorragias intracranianas, hemorragias gastrointestinais, equimoses e outras hemorragias) foi de 32,6% nos pacientes que receberam ruxolitinibe e 23,2% nos pacientes que receberam tratamento de controle (placebo ou tratamento ótimo disponível). A frequência de eventos de grau 3-4 foi semelhante nos pacientes que receberam ruxolitinibe (4,7%) e nos que receberam tratamento de controle (3,1%). A maioria dos pacientes com hemorragia relatou equimoses (65,3%). A frequência de equimoses foi maior nos pacientes que receberam ruxolitinibe (21,3%) em comparação com os que receberam tratamento de controle (11,6%). A frequência de hemorragias intracranianas foi de 1% nos pacientes que receberam ruxolitinibe e 0,9% nos pacientes que receberam tratamento de controle. A frequência de hemorragias gastrointestinais foi de 5,0% nos pacientes que receberam ruxolitinibe, em comparação com 3,1% nos pacientes que receberam tratamento de controle. Outras hemorragias (incluindo epistaxe, hemorragias pós-procedimento e hematúria) foram relatadas em 13,3% dos pacientes que receberam ruxolitinibe e 10,3% dos pacientes que receberam tratamento de controle.
Durante o acompanhamento de longo prazo nos estudos de fase 3, a frequência cumulativa de eventos de hemorragia aumentou proporcionalmente ao aumento do tempo de acompanhamento. A formação de equimoses foi o evento de hemorragia mais frequentemente relatado (33,3%). As hemorragias intracranianas e as hemorragias gastrointestinais ocorreram em 1,3% e 10,1% dos pacientes, respectivamente.
No período de comparação dos estudos de fase 3, a frequência de eventos de hemorragia (incluindo hemorragias intracranianas, hemorragias gastrointestinais, equimoses e outras hemorragias) foi de 16,8% nos pacientes que receberam ruxolitinibe, em comparação com 15,3% nos pacientes que receberam tratamento ótimo disponível no estudo RESPONSE e 12,0% nos pacientes que receberam tratamento ótimo disponível no estudo RESPONSE 2. A frequência de equimoses foi de 10,3% nos pacientes que receberam ruxolitinibe, em comparação com 8,1% nos pacientes que receberam tratamento ótimo disponível no estudo RESPONSE e 2,7% nos pacientes que receberam tratamento ótimo disponível no estudo RESPONSE 2. Não houve relatos de hemorragias intracranianas ou hemorragias gastrointestinais nos pacientes que receberam ruxolitinibe. Um paciente que recebeu ruxolitinibe teve um evento de hemorragia de grau 3 (hemorragia pós-procedimento); não houve relatos de eventos de hemorragia de grau 4. Outras hemorragias (incluindo epistaxe, hemorragias pós-procedimento e hematúria) foram relatadas em 8,7% dos pacientes que receberam ruxolitinibe, em comparação com 6,3% dos pacientes que receberam tratamento ótimo disponível no estudo RESPONSE e 6,7% dos pacientes que receberam tratamento ótimo disponível no estudo RESPONSE 2.
Durante o acompanhamento de longo prazo nos estudos de fase 3, a frequência cumulativa de eventos de hemorragia aumentou proporcionalmente ao aumento do tempo de acompanhamento. A formação de equimoses foi o evento de hemorragia mais frequentemente relatado (17,4%). As hemorragias intracranianas e as hemorragias gastrointestinais ocorreram em 0,3% e 3,5% dos pacientes, respectivamente.
No período de comparação do estudo de fase 3, a frequência de eventos de hemorragia foi de 25,0% e 22,0% nos pacientes que receberam ruxolitinibe e nos pacientes que receberam tratamento ótimo disponível, respectivamente. A frequência de eventos de hemorragia foi semelhante nos pacientes dos dois grupos de tratamento: equimoses (5,9% no grupo de ruxolitinibe versus 6,7% no grupo de tratamento ótimo disponível), hemorragias gastrointestinais (9,2% versus 6,7%) e outras hemorragias (13,2% versus 10,7%). As hemorragias intracranianas ocorreram em 0,7% dos pacientes no grupo de tratamento ótimo disponível e não foram relatadas em nenhum paciente no grupo de ruxolitinibe.
No período de comparação do estudo de fase 3, a frequência de eventos de hemorragia foi de 11,5% e 14,6% nos pacientes que receberam ruxolitinibe e nos pacientes que receberam tratamento ótimo disponível, respectivamente. A frequência de eventos de hemorragia foi semelhante nos pacientes dos dois grupos de tratamento: equimoses (4,2% no grupo de ruxolitinibe versus 2,5% no grupo de tratamento ótimo disponível), hemorragias gastrointestinais (1,2% versus 3,2%) e outras hemorragias (6,7% versus 10,1%). Não houve relatos de hemorragias intracranianas em nenhum dos grupos de tratamento.
Infecções
Nos estudos de fase 3, a frequência de infecção do trato urinário de grau 3 ou 4 foi de 1,0% nos pacientes que receberam ruxolitinibe, em comparação com 0,4% nos pacientes que receberam tratamento de controle. A frequência de herpes zóster foi semelhante nos pacientes com MF (4,0%) e PV (4,3%). Foi relatado um caso de neuropatia pós-herpética de grau 3, de acordo com a CTCAE, em pacientes com PV. A pneumonia foi relatada em 0,5% dos pacientes que receberam ruxolitinibe, em comparação com 1,6% dos pacientes que receberam tratamento de controle. Não houve relatos de sepse ou tuberculose nos pacientes que receberam ruxolitinibe.
Durante o acompanhamento de longo prazo nos estudos de fase 3, as infecções do trato urinário foram relatadas em 11,8% dos pacientes, herpes zóster em 14,7% e pneumonia em 7,1%. A sepse foi relatada em 0,6% dos pacientes. Não houve relatos de tuberculose durante o acompanhamento de longo prazo.
No período de comparação do estudo de fase 3, as infecções do trato urinário ocorreram em 9,9% (grau ≥ 3, 3,3%) dos pacientes no grupo de ruxolitinibe, em comparação com 10,7% (grau ≥ 3, 6,0%) no grupo de tratamento ótimo disponível. As infecções por citomegalovirus ocorreram em 28,3% (grau ≥ 3, 9,3%) dos pacientes no grupo de ruxolitinibe, em comparação com 24,0% (grau ≥ 3, 10,0%) no grupo de tratamento ótimo disponível. A sepse ocorreu em 12,5% (grau ≥ 3, 11,1%) dos pacientes no grupo de ruxolitinibe, em comparação com 8,7% (grau ≥ 3, 6,0%) no grupo de tratamento ótimo disponível. A infecção por vírus BK ocorreu apenas no grupo de ruxolitinibe, em 3 pacientes, com um caso de grau 3. Durante o acompanhamento de longo prazo, as infecções do trato urinário ocorreram em 17,9% (grau ≥ 3, 6,5%) dos pacientes, e as infecções por citomegalovirus ocorreram em 32,3% (grau ≥ 3, 11,4%) dos pacientes. As infecções por citomegalovirus com envolvimento de órgãos ocorreram em muito poucos pacientes; colite por citomegalovirus, enterite por citomegalovirus e infecção por citomegalovirus do trato gastrointestinal de qualquer grau ocorreram em 4, 2 e 1 paciente, respectivamente. A sepse de qualquer grau ocorreu em 25,4% (grau ≥ 3, 21,9%) dos pacientes.
No período de comparação do estudo de fase 3, as infecções do trato urinário ocorreram em 8,5% (grau ≥ 3, 1,2%) dos pacientes no grupo de ruxolitinibe, em comparação com 6,3% (grau ≥ 3, 1,3%) no grupo de tratamento ótimo disponível. A infecção por vírus BK ocorreu em 5,5% (grau ≥ 3, 0,6%) dos pacientes no grupo de ruxolitinibe, em comparação com 1,3% no grupo de tratamento ótimo disponível. As infecções por citomegalovirus ocorreram em 9,1% (grau ≥ 3, 1,8%) dos pacientes no grupo de ruxolitinibe, em comparação com 10,8% (grau ≥ 3, 1,9%) no grupo de tratamento ótimo disponível. A sepse ocorreu em 2,4% (grau ≥ 3, 2,4%) dos pacientes no grupo de ruxolitinibe, em comparação com 6,3% (grau ≥ 3, 5,7%) no grupo de tratamento ótimo disponível. Durante o acompanhamento de longo prazo, as infecções do trato urinário e a infecção por vírus BK ocorreram em 9,3% (grau ≥ 3, 1,3%) e 4,9% (grau ≥ 3, 0,4%) dos pacientes, respectivamente. As infecções por citomegalovirus e a sepse ocorreram em 8,8% (grau ≥ 3, 1,3%) e 3,5% (grau ≥ 3, 3,5%) dos pacientes, respectivamente.
Aumento da Lipase
No período randomizado do estudo RESPONSE, a redução dos níveis de lipase foi maior no grupo de ruxolitinibe em comparação com o grupo de controle, principalmente devido à diferença nos aumentos de grau 1 (18,2% versus 8,1%). Os aumentos de grau ≥ 2 foram semelhantes nos grupos de tratamento. No estudo RESPONSE 2, a frequência foi comparável nos grupos de ruxolitinibe e controle (10,8% versus 8%). Durante o acompanhamento de longo prazo nos estudos de fase 3, 7,4% e 0,9% dos pacientes relataram aumentos de grau 3 e 4 na lipase, respectivamente. Nenhum desses pacientes relatou sinais ou sintomas de pancreatite ou aumento da lipase.
Nos estudos de fase 3, os níveis elevados de lipase foram relatados em 18,7% e 19,3% dos pacientes no grupo de ruxolitinibe, em comparação com 16,6% e 14,0% nos grupos de controle, nos estudos COMFORT I e COMFORT II, respectivamente. Não houve relatos de sinais ou sintomas de pancreatite em pacientes com níveis elevados de lipase.
No período de comparação do estudo de fase 3, novos ou piorados valores de lipase foram relatados em 19,7% dos pacientes no grupo de ruxolitinibe, em comparação com 12,5% no grupo de tratamento ótimo disponível; o aumento correspondente nos eventos de grau 3 (3,1% versus 5,1%) e grau 4 (0% versus 0,8%) foi semelhante. Durante o acompanhamento de longo prazo, os valores de lipase aumentados foram relatados em 32,2% dos pacientes; os eventos de grau 3 e 4 ocorreram em 8,7% e 2,2% dos pacientes, respectivamente.
No período de comparação do estudo de fase 3, novos ou piorados valores de lipase foram relatados em 32,1% dos pacientes no grupo de ruxolitinibe, em comparação com 23,5% no grupo de tratamento ótimo disponível; o aumento correspondente nos eventos de grau 3 (10,6% versus 6,2%) e grau 4 (0,6% versus 0%) foi semelhante. Durante o acompanhamento de longo prazo, os valores de lipase aumentados foram relatados em 35,9% dos pacientes; os eventos de grau 3 e 4 ocorreram em 9,5% e 0,4% dos pacientes, respectivamente.
Aumento da Pressão Arterial Sistólica
Nos estudos de fase 3, o aumento da pressão arterial sistólica em relação ao valor basal em 20 mmHg ou mais em pelo menos uma visita foi documentado em 31,5% dos pacientes, em comparação com 19,5% dos pacientes que receberam tratamento de controle. No estudo COMFORT I, a média do aumento da pressão arterial sistólica em relação ao valor basal foi de 0-2 mmHg no grupo de ruxolitinibe, em comparação com uma redução de 2-5 mmHg no grupo placebo. Os resultados do estudo COMFORT II mostraram uma diferença mínima nas médias entre os pacientes com MF que receberam ruxolitinibe e aqueles que receberam tratamento de controle.
No período randomizado do estudo de fase 3, a média do aumento da pressão arterial sistólica foi de 0,65 mmHg no grupo de ruxolitinibe, em comparação com uma redução de 2 mmHg no grupo de tratamento ótimo disponível.
Crianças
Um total de 20 pacientes com idade entre 12 e < 18 anos com TRT foi analisado para segurança: 9 pacientes (5 no grupo de ruxolitinibe e 4 no grupo de tratamento ótimo disponível) no estudo REACH2 e 11 pacientes (4 no grupo de ruxolitinibe e 7 no grupo de tratamento ótimo disponível) no estudo REACH3. Considerando a exposição semelhante observada em crianças e adultos, as reações adversas ao ruxolitinibe com a dose recomendada de 10 mg duas vezes ao dia são semelhantes em frequência e gravidade.
Pacientes Idosos
Um total de 29 pacientes no estudo REACH2 e 25 pacientes no estudo REACH3 com idade > 65 anos que receberam ruxolitinibe foi analisado para segurança. Não foram identificados novos problemas de segurança, e o perfil de segurança em pacientes com idade > 65 anos é geralmente consistente com o perfil de segurança observado em pacientes com idade entre 18 e 65 anos.
Notificação de Suspeitas de Reações Adversas
A notificação de reações adversas após a aprovação do medicamento é importante. Isso permite monitorar a relação benefício-risco do medicamento. Os profissionais de saúde e os pacientes ou seus representantes legais devem notificar todas as suspeitas de reações adversas e falta de eficácia do medicamento por meio do Sistema de Notificação de Reações Adversas.
Prazo de Validade
3 anos.
Condições de Armazenamento
Armazenar a uma temperatura não superior a 25°C. Armazenar em local inacessível a crianças.
Embalação
14 comprimidos em blister, 4 blisters em caixa de cartão.
Classificação de Venda
Somente com receita médica.
Fabricante
Novartis Pharma Stein AG.
Endereço do Fabricante
Schaffhauserstrasse, 4332 Stein, Suíça.
- País de registo
- Requer receita médicaSim
- Fabricante
- Esta informação é apenas para referência e não constitui aconselhamento médico. Consulte sempre um médico antes de tomar qualquer medicamento. A Oladoctor não se responsabiliza por decisões médicas baseadas neste conteúdo.
- Alternativas a VERRUKUTANForma farmacêutica: solução, 2% em frasco de 60 mlSubstância ativa: minoxidilNão requer receita médicaForma farmacêutica: solução, 5% em frasco de 60 mlSubstância ativa: minoxidilNão requer receita médicaForma farmacêutica: creme, 1%; 15g, 30g, 60g ou 100g em um tuboSubstância ativa: pimecrolimusFabricante: МЕДА МеньюфекчерингRequer receita médica
Alternativas a VERRUKUTAN noutros países
As melhores alternativas com o mesmo princípio ativo e efeito terapêutico.
Alternativa a VERRUKUTAN em Польща
Alternativa a VERRUKUTAN em Іспанія
Médicos online para VERRUKUTAN
Avaliação de posologia, efeitos secundários, interações, contraindicações e renovação da receita de VERRUKUTAN – sujeita a avaliação médica e regras locais.








Receba novidades da plataforma e promoções exclusivas
Fique a par das atualizações da Oladoctor e receba promoções exclusivas para subscritores.

