


Obizur 500 U pó e solvente para solução injetável
Pergunte a um médico sobre a prescrição de Obizur 500 U pó e solvente para solução injetável



Como usar Obizur 500 U pó e solvente para solução injetável
Introdução
Prospecto: informação para o utilizador
OBIZUR500U pó e dissolvente para solução injetável
Susoctocog alfa
Este medicamento está sujeito a monitorização adicional, o que agilizará a detecção de nova informação sobre a sua segurança. Pode contribuir comunicando os efeitos adversos que possa ter. A parte final da secção 4 inclui informação sobre como comunicar estes efeitos adversos.
Leia todo o prospecto atentamente antes de começar a usar este medicamento, porque contém informação importante para si.
- Conserva este prospecto, porque pode ter que voltar a lê-lo.
- Se tiver alguma dúvida, consulte o seu médico.
- Este medicamento foi prescrito apenas para si, e não deve dá-lo a outras pessoas, mesmo que tenham os mesmos sintomas que si, porque pode prejudicá-las.
- Se experimentar efeitos adversos, consulte o seu médico, mesmo que se trate de efeitos adversos que não aparecem neste prospecto. Ver secção 4.
Conteúdo do prospecto
- O que é OBIZUR e para que é utilizado
- O que precisa saber antes de começar a usar OBIZUR
- Como usar OBIZUR
- Efeitos adversos possíveis
- Conservação de OBIZUR
- Conteúdo do envase e informação adicional
1. O que é OBIZUR e para que é utilizado
OBIZUR contém o princípio ativo susoctocog alfa, fator VIII antihemofílico, sequência porcina. O fator VIII é necessário para que o sangue forme coágulos e para deter os sangramentos.
Nos pacientes com hemofilia adquirida, o FVIII não funciona corretamente porque o paciente produziu anticorpos contra o seu próprio fator VIII que neutralizam este fator da coagulação sanguínea.
OBIZUR é utilizado para o tratamento dos episódios de sangramento em adultos com hemofilia adquirida (um distúrbio hemorrágico causado pela falta de atividade de fator VIII devido à produção de anticorpos). O efeito neutralizador destes anticorpos face a OBIZUR é menor do que face ao fator VIII humano.
OBIZUR restabelece a atividade de fator VIII ausente e ajuda a que o sangue forme coágulos no local do sangramento.
2. O que precisa saber antes de começar a usar OBIZUR
O produto só pode ser administrado a pacientes hospitalizados, porque é necessário supervisionar clinicamente o estado hemorrágico do paciente.
Não use OBIZUR:
- se é alérgico ao susoctocog alfa ou a algum dos outros componentes deste medicamento (incluídos na secção 6)
- se é alérgico a proteínas de hámster (OBIZUR pode conter quantidades mínimas derivadas do processo de fabricação)
Em caso de dúvida, consulte o seu médico antes de começar a usar este medicamento.
Advertências e precauções
Consulte o seu médico antes de começar a usar OBIZUR.
Existe uma possibilidade muito pequena de que sofra uma reação alérgica a OBIZUR. Deve estar atento aos sinais iniciais das reações alérgicas (ver secção 4 para consultar os sinais e sintomas). Se aparecer algum destes sintomas, deve parar a injeção. Os sintomas graves, como a dificuldade para respirar e o (pré)desmaio, necessitam de tratamento urgente.
Pacientes que produzem anticorpos inibidores contra OBIZUR
O seu médico pode verificar se tem anticorpos inibidores contra o fator VIII porcino.
O seu médico verificará o fator VIII no sangue para confirmar que está a ser administrado suficiente fator VIII. O seu médico também verificará se o sangramento parou de forma satisfatória.
Informa ao seu médico se teve uma doença cardiovascular no passado ou se tem risco conhecido de trombose (doenças produzidas por coágulos de sangue nos vasos sanguíneos normais), porque não se pode descartar a possibilidade de sofrer doenças tromboembólicas com a administração de concentrações altas e prolongadas de fator VIII.
Nome e número do lote
Recomendamos encarecidamente que o profissional de saúde registe o nome e o número do lote do medicamento cada vez que use OBIZUR, com o fim de manter um vínculo entre o tratamento e o lote do medicamento.
Crianças e adolescentes
OBIZUR não está autorizado atualmente para o tratamento de pacientes menores de 18 anos de idade, nos quais a hemofilia adquirida é rara.
Uso de OBIZUR com outros medicamentos
Informa ao seu médico se está a usar, usou recentemente ou possa ter que usar qualquer outro medicamento. Não se conhecem interações entre OBIZUR e outros medicamentos.
Gravidez e amamentação
Se está grávida ou em período de amamentação, acha que possa estar grávida ou tem intenção de engravidar, consulte o seu médico antes de usar este medicamento.
Condução e uso de máquinas
OBIZUR não influencia a capacidade para conduzir e utilizar máquinas.
OBIZUR contém sódio
Este medicamento contém 4,4 mg de sódio por mililitro uma vez preparado.
Consulte o seu médico se segue uma dieta pobre em sódio.
3. Como usar OBIZUR
O tratamento com OBIZUR será realizado por um médico com experiência na atenção de pacientes com hemofilia (distúrbios hemorrágicos).
O seu médico calculará a dose de OBIZUR (em unidades ou U) dependendo do seu estado e peso corporal. A frequência e a duração da administração dependerão do grau de eficácia que OBIZUR tenha no seu caso. Normalmente, o tratamento substitutivo com OBIZUR é temporário até que desapareça o sangramento ou se erradiquem os anticorpos contra o seu próprio fator VIII.
A dose de início recomendada é de 200 U por quilograma de peso corporal administradas por injeção intravenosa.
O seu médico analisará a atividade de fator VIII cada certo tempo para decidir a próxima dose e frequência de OBIZUR.
É habitual que o sangramento remita nas primeiras 24 horas; o seu médico ajustará a dose e a duração de OBIZUR até que pare de sangrar.
O volume total de OBIZUR reconstituído deve ser administrado a uma velocidade de entre 1 e 2 ml por minuto.
Siga exatamente as instruções de administração deste medicamento indicadas pelo seu médico. Em caso de dúvida, consulte novamente o seu médico.
Se usar mais OBIZUR do que deve
Siga exatamente as instruções de administração de OBIZUR indicadas pelo seu médico. Se usar mais OBIZUR do que o recomendado, informe o seu médico o mais rápido possível.
Se esquecer de usar OBIZUR
Não use uma dose dupla para compensar as doses esquecidas. Consulte o seu médico se esqueceu de uma dose e não sabe como compensá-la.
Se interromper o tratamento com OBIZUR
Não interrompa o tratamento com OBIZUR sem consultar o seu médico.
Se tiver alguma outra dúvida sobre o uso deste medicamento, pergunte ao seu médico.
4. Efeitos adversos possíveis
Assim como todos os medicamentos, este medicamento pode produzir efeitos adversos, embora nem todas as pessoas os sofram.
Se ocorrerem reações alérgicas graves e repentinas, deve suspender a injeção imediatamente. Contacte o seu médico imediatamente se tiver algum dos seguintes sintomas iniciais:
- Inchaço dos lábios e da língua
- Coceira e picada no local de injeção
- Calafrios, rubor
- Urticária generalizada, coceira
- Dor de cabeça, pressão arterial baixa
- Letargia (sonolência), sensação de doença, inquietude
- Palpitações, opressão no peito
- Formigamento, vômitos
- Som silbante que se produz ao respirar (sibilância)
Efeitos adversos frequentes (podem afetar até 1 pessoa em cada 10)
- Produção de anticorpos contra o medicamento
Comunicação de efeitos adversos
Se experimentar qualquer tipo de efeito adverso, consulte o seu médico, mesmo que se trate de possíveis efeitos adversos que não aparecem neste prospecto. Também pode comunicá-los diretamente através do sistema nacional de notificação incluído no Apêndice V. Mediante a comunicação de efeitos adversos, pode contribuir para fornecer mais informação sobre a segurança deste medicamento.
5. Conservação de OBIZUR
Mantenha este medicamento fora da vista e do alcance das crianças.
Não use este medicamento após a data de validade que aparece na caixa, no frasco e na seringa pré-carregada após CAD. A data de validade é o último dia do mês que se indica.
Conservar em frigorífico (entre 2°C e 8°C).
Não congelar.
Use a solução reconstituída imediatamente e nunca mais de 3 horas após a dissolução completa do pó.
Após a reconstituição, a solução deve ser transparente e incolor.
Não a administre se detectar partículas ou mudança de cor.
Os medicamentos não devem ser jogados nos esgotos nem na lixeira. Pergunte ao seu farmacêutico como se livrar dos envases e dos medicamentos que já não precisa. Dessa forma, ajudará a proteger o meio ambiente.
6. Titular da autorização de comercialização e responsável pela fabricação
Composição de OBIZUR
- O princípio ativo é susoctocogo alfa (fator VIII antihemofílico, sequência porcina, produzido mediante tecnologia de DNA recombinante). Cada frasco de pó contém 500 U de susoctocogo alfa.
- Os demais componentes do pó são polissorbato 80, cloreto de sódio (ver também a seção 2), cloreto de cálcio diidratado, sacarose, Tris base, Tris HCl, citrato trissódico diidratado.
- O diluente é 1 ml de água esterilizada para preparações injetáveis.
Aspecto do produto e conteúdo do envase
Um envase contém 1, 5 ou 10 unidades do seguinte:
- frasco de vidro com 500 U de OBIZUR em forma de pó friável branco provido de tampão de borracha e cápsula de fechamento do tipo «flip-off»
- seringa de vidro pré-carregada com 1 ml de água esterilizada para preparações injetáveis provida de protetor de borracha butílica e adaptador para conexão Luer
- dispositivo de transferência de líquido com punção de plástico integrado
Título da autorização de comercialização e responsável pela fabricação
Título da autorização de comercialização
Baxalta Innovations GmbH
Industriestrasse 67
A-1221 Viena
Áustria
Responsável pela fabricação
Baxter AG
Industriestrasse 67
A-1221 Viena
Áustria
Podem solicitar mais informações sobre este medicamento dirigindo-se ao representante local do titular da autorização de comercialização:
Bélgica Baxalta Belgium SPRL Tel.: +32 2 892 62 00 | Lituânia UAB Baxter Lithuania Tel: +370 5 269 16 90 / +370 5 252 71 00 |
Bulgária Baxalta Bulgaria EOOD Tel.: +359 2 9808482 | Luxemburgo Baxalta Belgium SPRL Tel: +32 2 892 62 00 |
República Tcheca Baxter Czech spol.s r.o. Tel.: +420 225774111 | Hungria Baxter Hungary Kft Tel.: +36 1 202 1980 |
Dinamarca Baxalta Denmark A/S Tlf.: +45 32 70 12 00 | Malta Baxalta UK Limited Tel.: +44 1 635 798 777 |
Alemanha Baxalta Deutschland GmbH Tel.: +49 89 262077-011 | Países Baixos Baxalta Netherlands B.V. Tel.: +31 30 799 27 77 |
Estônia OÜ Baxter Estonia Tel.: +372 6 515 120 | Noruega Baxalta Norway AS Tlf.: +47 22 585 000 |
Grécia Baxter Hellas ΕΠΕ Τηλ.: +30 210 28 80 000 | Áustria Baxalta Österreich GmbH Tel.: +43 1 20100-0 |
Espanha Baxalta Spain S.L. Tel.: +34 91 790 42 22 | Polônia Baxter Polska Sp. z.o.o. Tel.: +48 22 4883 777 |
França Baxalta France SAS Tél.: +33 1 70 96 06 00 | Portugal Baxalta Portugal, Unipessoal, Lda. Tel.: +351 21 122 03 00 |
Croácia Baxter d.o.o. Tel.: +386 1 420 16 80 | Romênia FARMACEUTICA REMEDIA SA Tel.: +40 21 321 16 40 |
Irlanda Baxalta UK Limited Tel.: +44 1 635 798 777 | Eslovênia Baxter d.o.o. Tel.: +386 1 420 16 80 |
Islândia Lyfjaver ehf. Sími: +354 533 6100 | República Eslovaca Baxter Slovakia, s.r.o. Tel: +421 2 3210 1150 |
Itália Baxalta Italy S.r.l. Tel.: +39 06 45224 600 | Finlândia Baxalta Finland Oy Puh/Tel.: +358 201 478 200 |
Chipre Baxter Hellas ΕΠΕ Τηλ.: +30 210 28 80 000 | Suécia Baxalta Sweden AB Tel.: +46 8 50 53 26 00 |
Letônia SIA Baxter Latvia Tel.: +371 67 784 784 | Reino Unido Baxalta UK Limited Tel.: +44 1 635 798 777 |
Data da última revisão deste prospecto:
Este medicamento foi autorizado em «circunstâncias excepcionais». Esta modalidade de aprovação significa que devido à rareza da doença não foi possível obter informações completas sobre este medicamento.
A Agência Europeia de Medicamentos revisará anualmente as novas informações sobre este medicamento que possam estar disponíveis e este prospecto será atualizado quando necessário.
Outras fontes de informação
As informações detalhadas sobre este medicamento estão disponíveis no site da Agência Europeia de Medicamentos: http://www.ema.europa.eu, e no site da Agência Espanhola de Medicamentos e Produtos Sanitários (AEMPS) (http://www.aemps.gob.es/). Também existem links para outros sites sobre doenças raras e medicamentos órfãos.
No site da Agência Europeia de Medicamentos, pode encontrar este prospecto em todas as línguas da União Europeia/Espaço Econômico Europeu.
‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑
Esta informação é destinada apenas a profissionais do setor sanitário:
INSTRUÇÕES PARA A PREPARAÇÃO E ADMINISTRAÇÃO
Preparação
Antes de começar a reconstituição, você precisará do seguinte:
- Número calculado de frascos de pó
- Mesma quantidade de seringas de 1 ml de diluente e adaptadores para frasco estéreis
- Algodões impregnados de álcool
- Uma seringa estéril grande para introduzir o volume final do medicamento reconstituído
Os seguintes procedimentos são diretrizes gerais para a preparação e reconstituição de OBIZUR. Repita as seguintes instruções de reconstituição com cada frasco de pó que você vai reconstituir.
Reconstituição
Use uma técnica asséptica durante o procedimento de reconstituição.
- Deixe o frasco de pó de OBIZUR e a seringa pré-carregada de diluente atingirem a temperatura ambiente.
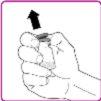
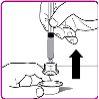
- Retire a cápsula de plástico do frasco de pó de OBIZUR (Figura A).
- Esfregue o tampão de borracha com um algodão impregnado em álcool (não incluído) e espere até que seque.
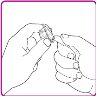
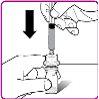
- Retire o protetor do envase do adaptador para frasco (Figura B). Não toque a conexão Luer localizada no centro do adaptador para frasco. Não retire o adaptador para frasco do envase.
- Coloque o envase com o adaptador para frasco em uma superfície limpa com a conexão Luer mirando para cima.
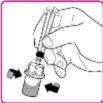
- Rompa o lacre inviolável da seringa pré-carregada de diluente (Figura C).
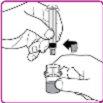
- Segure com firmeza o envase com o adaptador para frasco e conecte a seringa pré-carregada de diluente ao adaptador pressionando o cone da seringa contra a conexão Luer localizada no centro do adaptador e girando-o no sentido dos ponteiros do relógio até que a seringa esteja bem segura. Não aperte em excesso (Figura D).
- Retire o envase de plástico (Figura E).
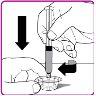
- Coloque o frasco de pó de OBIZUR em uma superfície limpa, plana e dura. Coloque o adaptador para frasco sobre o frasco de pó de OBIZUR e empurre com firmeza o punção com filtro do adaptador para frasco através do círculo de borracha do frasco de pó de OBIZUR até que o protetor de plástico transparente encaixe no frasco (Figura F).
- Empurre o êmbolo lentamente até injetar todo o diluente da seringa no frasco de pó de OBIZUR.
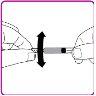
- Mova suavemente (com um movimento circular) o frasco de pó de OBIZUR sem retirar a seringa até que todo o pó se dissolva/reconstitua (Figura G). Antes de administrar a solução reconstituída, examine-a visualmente para verificar se não contém partículas. Não a use se observar partículas ou mudança de cor.
- Segure o frasco de pó e o adaptador para frasco com uma mão, agarre com firmeza o cilindro da seringa pré-carregada de diluente com a outra e desrosqueie a seringa do adaptador para frasco girando-a em sentido contrário ao dos ponteiros do relógio (Figura H).
- Se guardar OBIZUR a temperatura ambiente, use-o imediatamente e nas 3 horas seguintes à reconstituição.
Figura A | Figura B | Figura C | Figura D |
|
|
|
|
Figura E | Figura F | Figura G | Figura H |
|
|
|
|
Administração
Somente para injeção intravenosa!
- Antes de administrar a solução de OBIZUR reconstituída, examine-a visualmente para verificar se não contém partículas ou mudança de cor. A solução deve ser transparente e incolor a vista. Não a administre se observar partículas ou mudança de cor.
- Não administre OBIZUR no mesmo tubo ou envase que outros medicamentos injetáveis.
Usando uma técnica asséptica, administre a solução seguindo o seguinte procedimento:
- Uma vez reconstituídos todos os frascos, conecte uma seringa grande ao adaptador para frasco pressionando suavemente o cone da seringa contra a conexão Luer localizada no centro do adaptador e girando-o no sentido dos ponteiros do relógio até que a seringa esteja bem segura.
- Dê a volta ao frasco; expulse o ar da seringa no frasco e retire a solução de OBIZUR reconstituída para a seringa (Figura I).
Figura I
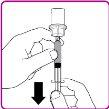
- Desrosqueie a seringa grande do adaptador para frasco girando-a em sentido contrário ao dos ponteiros do relógio e repita o processo com todos os frascos reconstituídos de OBIZUR até atingir o volume total que você vai administrar.
- Administre a solução de OBIZUR reconstituída por via intravenosa a uma velocidade de 1 a 2 ml por minuto.
A dose inicial de OBIZUR necessária para um paciente é calculada mediante a seguinte fórmula:
Dose inicial (U/kg) = concentração do produto (U/frasco) × peso corporal (kg) = número de frascos
por exemplo, o número de frascos para a dose inicial em um sujeito de 70 kg é calculado da seguinte forma:
200 U/kg = 500 U/frasco × 70 kg = 28 frascos
Dosagem
A dose inicial recomendada é de 200 U por quilograma de peso corporal, administrada por injeção intravenosa.
Tipo de sangramento | Atividade mínima de fator VIII desejada (Unidades por dl ou % do normal) | Dose inicial (Unidades por kg) | Dose seguinte | Frequência e duração das doses seguintes |
Sangramento leve a moderado do músculo superficial/sem afetação neurovascular e sangramento articular | > 50 % | 200 | Ajuste as doses seguintes com base na resposta clínica e para manter a atividade mínima desejada de fator VIII | Administre as doses a intervalos de 4 a 12 horas; a frequência pode ser ajustada com base na resposta clínica e na atividade de fator VIII quantificada |
Sangramento intramuscular, retroperitoneal, gastrointestinal, intracraniano importante de moderado a grave | > 80 % |
- País de registo
- Substância ativa
- Requer receita médicaSim
- Fabricante
- Esta informação é apenas para referência e não constitui aconselhamento médico. Consulte sempre um médico antes de tomar qualquer medicamento. A Oladoctor não se responsabiliza por decisões médicas baseadas neste conteúdo.
- Alternativas a Obizur 500 U pó e solvente para solução injetávelForma farmacêutica: INJETÁVEL, 1.000 UISubstância ativa: coagulation factor VIIIFabricante: Takeda Manufacturing Austria AgRequer receita médicaForma farmacêutica: INJETÁVEL, 1500 UISubstância ativa: coagulation factor VIIIFabricante: Takeda Manufacturing Austria AgRequer receita médicaForma farmacêutica: INJETÁVEL, 1000 UI - após reconstituição em 2 ml de água para injetáveis, a dose é de 500 UI/mlSubstância ativa: coagulation factor VIIIFabricante: Takeda Manufacturing Austria AgRequer receita médica
Médicos online para Obizur 500 U pó e solvente para solução injetável
Avaliação de posologia, efeitos secundários, interações, contraindicações e renovação da receita de Obizur 500 U pó e solvente para solução injetável – sujeita a avaliação médica e regras locais.









Receba novidades da plataforma e promoções exclusivas
Fique a par das atualizações da Oladoctor e receba promoções exclusivas para subscritores.

