

LUXTURNA 5 x 10¹² UNIDADES DE VETOR GENÉTICO/ML CONCENTRADO E SOLVENTE PARA SOLUÇÃO INJETÁVEL
Pergunte a um médico sobre a prescrição de LUXTURNA 5 x 10¹² UNIDADES DE VETOR GENÉTICO/ML CONCENTRADO E SOLVENTE PARA SOLUÇÃO INJETÁVEL



Como usar LUXTURNA 5 x 10¹² UNIDADES DE VETOR GENÉTICO/ML CONCENTRADO E SOLVENTE PARA SOLUÇÃO INJETÁVEL
Introdução
Prospecto: informação para o paciente
Luxturna 5× 1012genomas vetoriais/ml concentrado e diluente para solução injetável
voretigén neparvovec
Este medicamento está sujeito a monitorização adicional, o que agilizará a detecção de nova informação sobre a sua segurança. Pode contribuir comunicando os efeitos adversos que possa ter. A parte final da seção 4 inclui informação sobre como comunicar estes efeitos adversos.
Leia todo o prospecto atentamente antes de ser administrado este medicamento, porque contém informação importante para si.
- Conserva este prospecto, porque pode ter que voltar a lê-lo.
- Se tiver alguma dúvida, consulte o seu médico ou enfermeiro.
- Se experimentar efeitos adversos, consulte o seu médico ou enfermeiro, mesmo que se trate de efeitos adversos que não aparecem neste prospecto. Ver seção 4.
Conteúdo do prospecto:
- O que é Luxturna e para que é utilizado
- O que precisa saber antes de começar a receber Luxturna
- Como é administrado Luxturna
- Posíveis efeitos adversos
- Como conservar Luxturna
- Conteúdo do envase e informação adicional
1. O que é Luxturna e para que é utilizado
Luxturna é um produto de terapia génica que contém a substância ativa voretigén neparvovec.
Luxturna é utilizado para o tratamento de adultos e crianças com perda de visão devido a uma distrofia retiniana hereditária causada por mutações no gene RPE65.Estas mutações evitam que o corpo produza uma proteína necessária para a visão e conduzem a uma perda de vista e a uma possível cegueira.
O princípio ativo de Luxturna, voretigén neparvovec, é um vírus modificado que contém uma cópia do gene RPE65. Depois da injeção, este gene chega às células da retina, a camada da parte posterior do olho que detecta a luz. Isso permite que a retina produza as proteínas necessárias para a visão. O vírus utilizado para administrar o gene não causa doença em humanos.
Apenas lhe será administrado Luxturna se os testes genéticos mostrarem que a sua perda de visão é causada por mutações no gene RPE65.
2. O que precisa saber antes de começar a receber Luxturna
Não deve receber Luxturna
- Se é alérgico a voretigén neparvovec ou a algum dos outros componentes deste medicamento (incluídos na seção 6)
- Se tem uma infecção no olho
- Se tem uma inflamação no olho
Se está afetado por alguma das situações acima, ou se não está seguro, consulte o seu médico antes de ser administrado Luxturna.
Advertências e precauções
Antes de receber o tratamento com Luxturna:
- Informa ao seu médico se tem sinais de infecção ocular ou inflamação ocular, por exemplo se tem vermelhidão nos olhos, sensibilidade à luz, inchaço ocular ou dor nos olhos.
- Informa ao seu médico se tem uma infecção ativa de qualquer tipo. O seu médico pode adiar o seu tratamento até que a infecção desapareça porque este medicamento pode dificultar a luta contra a infecção. Ver também a seção 3.
Depois de receber o tratamento com Luxturna:
- Consulte imediatamente o seu médico se o seu olho/olhos se tornam vermelhos, se sente dor nos olhos, sensibilidade à luz, vê faíscas ou corpos flutuantes, ou se nota um agravamento ou visão borrosa.
- Deve evitar viagens de avião ou outros viagens a alturas elevadas até que o seu médico o indique. Durante o tratamento com este medicamento, o médico insere uma bolha de ar no olho, que o seu corpo absorve lentamente. Até que a bolha seja completamente absorvida, a viagem de avião ou outro viagem a alturas elevadas pode causar um crescimento da bolha e provocar danos nos olhos, incluindo a perda de visão. Consulte o seu médico antes de viajar.
- Deve evitar nadar devido a um maior risco de infecção no olho. Consulte o seu médico antes de ir nadar após receber o tratamento com Luxturna.
- Deve evitar a atividade física extenuante devido a um maior risco de lesão no olho. Consulte o seu médico antes de começar a realizar uma atividade física extenuante após receber o tratamento com Luxturna.
- Pode ter alterações visuais transitórias, como sensibilidade à luz e visão borrosa. Informe ao seu médico sobre qualquer alteração visual que experimente. O seu médico pode ajudá-lo a reduzir qualquer desconforto causado por estas alterações transitórias.
- O princípio ativo de Luxturna pode ser excretado temporariamente pelas lágrimas. Você e o seu cuidador devem colocar os curativos e o material de descarte que esteve em contacto com lágrimas e secreções nasais em sacos selados antes de descartá-los. Deve seguir estas precauções durante 14 dias.
- Não poderá doar sangue, órgãos, tecidos e células para transplante após ter sido tratado com Luxturna.
Crianças e adolescentes
Luxturna não foi estudado em crianças menores de quatro anos de idade. Os dados são limitados.
Outros medicamentos e Luxturna
Informa ao seu médico se está tomando, tomou recentemente ou possa ter que tomar qualquer outro medicamento.
Gravidez, lactação e fertilidade
Se está grávida ou em período de lactação, acredita que possa estar grávida ou tem intenção de engravidar, consulte o seu médico ou enfermeiro antes de receber o tratamento com Luxturna.
Os efeitos deste medicamento sobre a gravidez e o feto são desconhecidos. Como precaução, não deve receber Luxturna enquanto está grávida.
Luxturna não foi estudado em mulheres em período de lactação. Não se sabe se passa para o leite materno. Informe ao seu médico se está amamentando ou tivera planeado fazer isso. O seu médico o ajudará a decidir se tem que interromper a lactação ou não receber Luxturna, tendo em conta os benefícios da lactação no seu bebê e os benefícios de Luxturna em você.
Condução e uso de máquinas
Pode ter alterações visuais transitórias após receber Luxturna. Não conduza nem use máquinas pesadas até que a sua visão se tenha recuperado. Consulte o seu médico antes de retomar estas atividades.
Luxturna contém sódio
Este medicamento contém menos de 1 mmol de sódio (23 mg) por dose, isto é, essencialmente "isento de sódio".
3. Como é administrado Luxturna
Luxturna será administrado em uma sala de operações por cirurgiões com experiência na realização de cirurgia ocular. Luxturna é administrado sob anestesia. O seu médico falará com você sobre a anestesia e sobre como será administrado.
O seu médico realizará uma cirurgia ocular para eliminar o gel transparente que ocupa o interior do olho, e luego injetará Luxturna diretamente na retina, a camada fina sensível à luz que se encontra na parte posterior do olho. Este procedimento será repetido no outro olho pelo menos 6 dias depois. Terá que ficar em observação pós-operatória durante algumas horas após cada procedimento para controlar a sua recuperação e observar os efeitos secundários da cirurgia ou da anestesia.
Antes de iniciar o tratamento com Luxturna, o seu médico pode pedir-lhe que tome um medicamento que suprima o seu sistema imunológico (as defesas naturais do corpo) para que não tente lutar contra Luxturna quando for administrado. É importante que tome este medicamento de acordo com as instruções que o seu médico indicar. Não deixe de tomar o medicamento sem consultar primeiro o seu médico.
Se receber mais Luxturna do que deve
Como este medicamento será administrado por um médico, é pouco provável que seja administrado mais medicamento do que deve. Se isso ocorrer, o seu médico tratará os sintomas conforme necessário. Informe ao seu médico ou enfermeiro se tiver algum problema de visão.
Se tiver alguma outra dúvida sobre o uso deste medicamento, pergunte ao seu médico ou enfermeiro.
4. Posíveis efeitos adversos
Como todos os medicamentos, este medicamento pode produzir efeitos adversos, embora nem todas as pessoas os sofram.
Podem aparecer os seguintes efeitos adversos relacionados com Luxturna:
Frequentes (podem afetar até 1de cada 10pessoas)
- Depósitos debaixo da retina
Frequência não conhecida (não pode ser estimada a partir dos dados disponíveis)
- Atrofia da (corio)retina
Podem aparecer os seguintes efeitos adversos relacionados com o procedimento de injeção:
Muito frequentes(podem afetar mais de 1de cada 10pessoas)
- Vermelhidão nos olhos
- Catarata (opacidade do cristalino)
- Aumento da pressão no olho
Frequentes (podem afetar até 1de cada 10pessoas)
- Desgarro na retina
- Dor nos olhos
- Inchaço nos olhos
- Desprendimento da retina
- Sangramento na parte posterior do olho
- Dor ou aumento das molestias no olho
- Visão borrosa devido a um buraco na retina
- Adelgaçamento da superfície do olho (dellen)
- Irritação ocular
- Inflamação ocular
- Sensação de corpo estranho no olho
- Molestias oculares
- Anomalias na parte posterior do olho
- Náuseas (vontade de vomitar), vómitos, dor abdominal (estomacal), dor nos lábios
- Mudança na atividade elétrica do coração
- Dor de cabeça, tonturas
- Erupção cutânea, inchaço da face
- Ansiedade
- Problemas associados com a colocação de um tubo de respiração na traqueia
- Rotura da ferida quirúrgica
Não conhecidos (a frequência não pode ser estimada a partir dos dados disponíveis)
- Enturbiação da substância gelatinosa que se encontra no interior do olho (opacidades vítreas)
- Atrofia da (corio)retina
O dano dos tecidos do olho pode ir acompanhado de sangramento, de inflamação e de um maior risco de infecção. Produz-se uma redução da visão nos dias posteriores à cirurgia que geralmente melhora; informe ao seu médico se a visão não regressa.
Comunicação de efeitos adversos
Se experimentar qualquer tipo de efeito adverso, consulte o seu médico ou enfermeiro, mesmo que se trate de possíveis efeitos adversos que não aparecem neste prospecto. Também pode comunicá-los diretamente através do sistema nacional de notificação incluído no Apêndice V. Mediante a comunicação de efeitos adversos, você pode contribuir para proporcionar mais informação sobre a segurança deste medicamento.
5. Conservação de Luxturna
Luxturna será conservado pelos profissionais de saúde no seu centro de saúde.
O concentrado e o diluente devem ser transportados e armazenados congelados a temperatura ≤ ‑65 ºC. Uma vez descongelado, o medicamento não deve voltar a ser congelado e deve ser deixado à temperatura ambiente (por debaixo de 25 °C).
Não utilize este medicamento após a data de validade que aparece na etiqueta e na caixa após CAD.
6. Conteúdo do envase e informação adicional
Composição de Luxturna
- O princípio ativo é voretigén neparvovec. Cada ml de concentrado contém 5 × 10^12 genomas vectoriais (vg). O concentrado (frasco unidose de 2 ml com volume extráível de 0,5 ml) requer uma diluição de 1:10 antes da administração.
- Cada dose de solução diluída contém 1,5 × 10^11 genomas vectoriais de voretigén neparvovec em um volume administrável de 0,3 ml.
- Os demais excipientes do concentrado são cloreto de sódio (ver "Luxturna contém sódio" no final da seção 2 deste prospecto), dihidrogenofosfato de sódio monohidrato (para ajustar o pH), dihidrogenofosfato de sódio dihidrato (para ajustar o pH), poloxâmero 188 e água para preparações injetáveis.
- O diluente contém cloreto de sódio (ver o final da seção 2), dihidrogenofosfato de sódio monohidrato (para ajustar o pH), dihidrogenofosfato de sódio dihidrato (para ajustar o pH), poloxâmero 188 e água para preparações injetáveis.
Este medicamento contém organismos modificados geneticamente.
Aspecto de Luxturna e conteúdo do envase
Luxturna é um concentrado claro e incolor para solução para injeção subretiniana que se apresenta em um frasco de plástico transparente. O diluente é um líquido transparente e incolor que se apresenta em um frasco de plástico transparente.
Cada bolsa de alumínio contém uma caixa de cartão que inclui 1 frasco de 0,5 ml de concentrado e 2 frascos de diluente (cada um contendo 1,7 ml).
Título da autorização de comercialização
Novartis Europharm Limited
Edifício Vista
Elm Park, Merrion Road
Dublin 4
Irlanda
Responsável pela fabricação
Novartis Pharma GmbH
Roonstrasse 25
90429 Nuremberga
Alemanha
Novartis Pharma GmbH
Sophie-Germain-Strasse 10
90443 Nuremberga
Alemanha
Pode solicitar mais informações sobre este medicamento dirigindo-se ao representante local do título da autorização de comercialização:
Bélgica Novartis Pharma N.V. Tel: +32 2 246 16 11 | Lituânia SIA Novartis Baltics Lietuvos filialas Tel: +370 5 269 16 50 |
| Luxemburgo Novartis Pharma N.V. Tel: +32 2 246 16 11 |
República Checa Novartis s.r.o. Tel: +420 225 775 111 | Hungria Novartis Hungária Kft. Tel: +36 1 457 65 00 |
Dinamarca Novartis Healthcare A/S Tel: +45 39 16 84 00 | Malta Novartis Pharma Services Inc. Tel: +356 2122 2872 |
Alemanha Novartis Pharma GmbH Tel: +49 911 273 0 | Países Baixos Novartis Pharma B.V. Tel: +31 88 04 52 111 |
Estônia SIA Novartis Baltics Eesti filiaal Tel: +372 66 30 810 | Noruega Novartis Norge AS Tel: +47 23 05 20 00 |
Grécia Novartis (Hellas) A.E.B.E. Tel: +30 210 281 17 12 | Áustria Novartis Pharma GmbH Tel: +43 1 86 6570 |
Espanha Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 | Polônia Novartis Poland Sp. z o.o. Tel: +48 22 375 4888 |
França Novartis Pharma S.A.S. Tel: +33 1 55 47 66 00 | Portugal Novartis Farma - Produtos Farmacêuticos, S.A. Tel: +351 21 000 8600 |
Croácia Novartis Hrvatska d.o.o. Tel: +385 1 6274 220 | Romênia Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
Irlanda Novartis Ireland Limited Tel: +353 1 260 12 55 | Eslovênia Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
Islândia Vistor hf. Tel: +354 535 7000 | República Eslovaca Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
Itália Novartis Farma S.p.A. Tel: +39 02 96 54 1 | Finlândia Novartis Finland Oy Tel: +358 (0)10 6133 200 |
Chipre Novartis Pharma Services Inc. Tel: +357 22 690 690 | Suécia Novartis Sverige AB Tel: +46 8 732 32 00 |
Letônia SIA Novartis Baltics Tel: +371 67 887 070 |

Data da última revisão deste prospecto:
Outras fontes de informação
Este prospecto está disponível em formato de arquivo de áudio e em um tamanho de letra grande na página web: http://www.voretigeneneparvovec.support
A informação detalhada deste medicamento está disponível na página web da Agência Europeia de Medicamentos: http://www.ema.europa.eu.
------------------------------------------------------------------------------------------------------------------------
Esta informação está destinada apenas a profissionais de saúde:
Precauções que devem ser tomadas antes de manipular ou administrar o medicamento
Este medicamento contém organismos modificados geneticamente. Deve ser usado um equipamento de proteção pessoal (que inclua bata de laboratório, óculos de segurança e luvas) enquanto se prepara ou administra voretigén neparvovec.
A pressão intraocular deve ser controlada adequadamente e deve ser monitorizada antes e depois da administração do medicamento.
Depois da administração, deve ser indicado aos pacientes que notifiquem imediatamente qualquer sintoma que sugira endoftalmite ou desprendimento de retina e devem ser tratados adequadamente.
Preparação prévia à administração
Cada embalagem que contém 1 frasco de concentrado e 2 frascos de diluente é para uso único.
Luxturna deve ser inspecionado visualmente antes da administração. Se forem detectadas partículas, turbidez ou decoloração, não deve ser usado o frasco unidose.
A preparação de Luxturna deve ser realizada dentro das 4 horas anteriores ao início do procedimento de administração, em condições assépticas e de acordo com o seguinte procedimento recomendado.
Deixar descongelar a temperatura ambiente um frasco de dose única de concentrado e dois frascos de diluente. Uma vez descongelados os 3 frascos (1 frasco de concentrado e 2 frascos de diluente), deve ser iniciada a diluição. Inverter suavemente os frascos cinco vezes para misturar os conteúdos.
Inspecionar se há partículas visíveis ou qualquer anomalia. A aparição de qualquer anomalia ou de partículas visíveis deve ser notificada ao Titular da Autorização de Comercialização e o produto não deve ser utilizado.
Transferir 2,7 ml de diluente procedente dos dois frascos descongelados e dispensar com uma seringa de 3 ml em um frasco estéril de vidro vazio de 10 ml.
Para a diluição, extrair 0,3 ml de concentrado descongelado com uma seringa de 1 ml e adicionar ao frasco estéril de 10 ml que contém o diluente. Inverter suavemente o frasco pelo menos cinco vezes para uma mistura adequada. Inspecionar se há partículas visíveis. A solução diluída deve ser clara ou ligeiramente opalescente. Etiquetar o frasco de vidro de 10 ml que contém o concentrado diluído da seguinte forma: "Luxturna diluído".
Não devem ser preparadas as seringas se o frasco mostrar algum dano ou se forem observadas partículas visíveis. Preparar as seringas para injeção extrayendo 0,8 ml da solução diluída em uma seringa estéril de 1 ml. Repetir o mesmo procedimento para preparar uma seringa de reposto. As seringas cheias de produto devem ser transportadas até o quarto de operações em um contenedor designado para esse fim.
Medidas que devem ser adotadas em caso de exposição acidental
Deve ser evitada a exposição acidental. Deve ser seguida a normativa local sobre biossegurança para a preparação, administração e manuseio de voretigén neparvovec.
- Deve ser usado um equipamento de proteção pessoal (que inclua bata de laboratório, óculos de segurança e luvas) enquanto se manipula ou administra voretigén neparvovec.
- Deve ser evitada a exposição acidental a voretigén neparvovec, incluindo o contato com a pele, os olhos e as membranas mucosas. Deve ser coberta qualquer lesão antes de manipular este medicamento.
- Qualquer derramamento de voretigén neparvovec deve ser tratado com um agente virucida, como hipoclorito de sódio a 1%, e deve ser seco com materiais absorventes.
- Todos os materiais que possam ter entrado em contato com voretigén neparvovec (p. ex., frasco, seringa, agulha, gazes de algodão, luvas, máscaras ou bandagens) devem ser eliminados de acordo com a normativa local sobre biossegurança.
Exposição acidental
- Em caso de exposição ocupacional acidental (p. ex., por salpicaduras nos olhos ou nas membranas mucosas), deve ser enxaguado com água limpa durante pelo menos 5 minutos.
- Em caso de exposição na pele lesionada ou de lesão por punção com a agulha, deve ser limpo bem o local afetado com água e sabão e/ou com um desinfetante.
Precauções que devem ser tomadas na eliminação do medicamento
Este medicamento contém organismos modificados geneticamente. A eliminação do medicamento não utilizado e de todos os materiais que tenham estado em contato com ele será realizada de acordo com a normativa local de resíduos farmacêuticos.
Posologia
O tratamento deve ser iniciado e administrado por um cirurgião de retina com experiência em cirurgia macular.
Os pacientes receberão uma dose única de voretigén neparvovec de 1,5 × 10^11 genomas vectoriais em cada olho. Cada dose será administrada dentro do espaço subretiniano em um volume total de 0,3 ml. A administração deve ser realizada de forma individualizada em cada olho em dias separados dentro de um curto intervalo de, pelo menos, seis dias de diferença entre cada procedimento cirúrgico.
Esquema imunomodulador
Antes de iniciar o esquema imunomodulador e antes da administração de voretigén neparvovec, deve ser examinado o paciente para detectar sintomas de doença infecciosa ativa de qualquer natureza, e em caso de tal infecção, o início do tratamento deve ser adiado até após o paciente se ter recuperado.
Recomenda-se iniciar o esquema imunomodulador 3 dias antes da administração de voretigén neparvovec no primeiro olho, seguindo o calendário descrito a seguir (Tabela 1). O início do esquema imunomodulador para o segundo olho deve seguir o mesmo esquema e deve substituir o esquema imunomodulador do primeiro olho.
Tabela1Esquema imunomodulador pré e pós-operatório para cada olho
Pré-operatório | 3 dias antes da administração de Luxturna | Prednisona (ou equivalente) 1 mg/kg/dia (até um máximo de 40 mg/dia) |
Pós-operatório | 4 dias (incluindo o dia da administração) | Prednisona (ou equivalente) 1 mg/kg/dia (até um máximo de 40 mg/dia) |
Continuar 5 dias | Prednisona (ou equivalente) 0,5 mg/kg/dia (até um máximo de 20 mg/dia) | |
Continuar 5 dias com uma dose a cada dois dias | Prednisona (ou equivalente) 0,5 mg/kg a cada dois dias (até um máximo de 20 mg/dia) |
Populações especiais
Pacientes idosos
Não foi estabelecida a segurança e eficácia de voretigén neparvovec em pacientes ≥ 65 anos. Os dados são limitados. No entanto, não é necessário um ajuste de dose em pacientes idosos.
Insuficiência hepática e renal
Não foi estabelecida a segurança e eficácia de voretigén neparvovec em pacientes com insuficiência hepática ou renal. Não é necessário um ajuste de dose nesses pacientes (ver seção 5.2).
População pediátrica
Não foi estabelecida a segurança e eficácia de voretigén neparvovec em crianças menores de 4 anos de idade. Os dados são limitados. Não é necessário um ajuste de dose em pacientes pediátricos.
Forma de administração
Uso subretiniano.
Luxturna é uma solução estéril concentrada para injeção subretiniana que requer descongelamento e diluição antes da administração.
Este medicamento não deve ser administrado por injeção intravítrea.
Luxturna é um frasco de uso único para uma administração única em um único olho. O produto é administrado por meio de uma injeção subretiniana após realizar uma vitrectomia em cada olho. Não deve ser administrado muito próximo à fóvea para manter a integridade foveal.
A administração de voretigén neparvovec deve ser realizada no quarto de operações sob condições assépticas controladas. Antes do procedimento, deve ser administrada ao paciente a anestesia adequada. A pupila do olho em que se vai administrar a injeção deve estar dilatada, e antes da cirurgia deve ser administrado um antibiótico de amplo espectro por via oftálmica de acordo com a prática médica habitual.
Administração
Sigam os passos descritos a seguir para administrar voretigén neparvovec aos pacientes:
- Uma vez diluído Luxturna, deve ser inspecionado visualmente antes da administração. Se forem observadas partículas, turbidez ou decoloração, o medicamento não deve ser utilizado.
- Conectar a seringa que contém o produto diluído ao tubo de extensão e à cânula de injeção subretiniana. O produto deve ser injetado lentamente através do tubo de extensão e da cânula de injeção subretiniana para eliminar qualquer bolha de ar no sistema.
- O volume de produto disponível para injeção é confirmado na seringa ao alinhar a ponta do êmbolo com a linha que marca 0,3 ml.
- Uma vez finalizada a vitrectomia, Luxturna é administrado por meio de injeção subretiniana utilizando uma cânula de injeção subretiniana introduzida por via pars plana.
- Sob visualização direta, a ponta da cânula de injeção subretiniana é colocada em contato com a superfície da retina. O local de injeção recomendado deve ser situado ao longo da arcada vascular superior, pelo menos a 2 mm de distância do centro da fóvea. É injetada lentamente uma pequena quantidade de produto até que seja observada uma ampola subretiniana inicial, e luego o volume restante é injetado lentamente até que sejam administrados os 0,3 ml totais (Figura 1).
Figura1Ponta da cânula de injeção subretiniana colocada no local de injeção recomendado (vista do cirurgião)
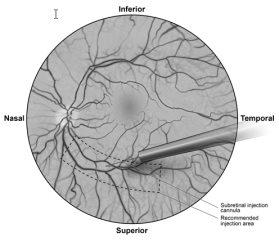
- Ao finalizar a injeção, a cânula de injeção subretiniana é retirada do olho.
- Depois da injeção, deve ser descartado qualquer produto não utilizado. Não deve ser guardada a seringa de reposto.
- Deve ser realizado cuidadosamente um intercâmbio fluido-ar, evitando o drenagem de líquido perto da retinotomia criada para a injeção subretiniana.
- No pós-operatório, a cabeça deve ser colocada em posição supina imediatamente e deve ser mantida durante 24 horas após a alta do paciente.
- País de registo
- Substância ativa
- Requer receita médicaSim
- Fabricante
- Esta informação é apenas para referência e não constitui aconselhamento médico. Consulte sempre um médico antes de tomar qualquer medicamento. A Oladoctor não se responsabiliza por decisões médicas baseadas neste conteúdo.
- Alternativas a LUXTURNA 5 x 10¹² UNIDADES DE VETOR GENÉTICO/ML CONCENTRADO E SOLVENTE PARA SOLUÇÃO INJETÁVELForma farmacêutica: COLÍRIO, 5,5 mg cloreto de sódio; 3 mg hipromelose/mlSubstância ativa: artificial tears and other indifferent preparationsFabricante: Alcon Healthcare S.A.Não requer receita médicaForma farmacêutica: COLÍRIO, 3,2 mg/mlSubstância ativa: artificial tears and other indifferent preparationsFabricante: Bausch & Lomb S.A.Não requer receita médicaForma farmacêutica: COLÍRIO, 3,2 mg/mlSubstância ativa: artificial tears and other indifferent preparationsFabricante: Bausch & Lomb S.A.Não requer receita médica
Alternativas a LUXTURNA 5 x 10¹² UNIDADES DE VETOR GENÉTICO/ML CONCENTRADO E SOLVENTE PARA SOLUÇÃO INJETÁVEL noutros países
As melhores alternativas com o mesmo princípio ativo e efeito terapêutico.
Alternativa a LUXTURNA 5 x 10¹² UNIDADES DE VETOR GENÉTICO/ML CONCENTRADO E SOLVENTE PARA SOLUÇÃO INJETÁVEL em Polónia
Alternativa a LUXTURNA 5 x 10¹² UNIDADES DE VETOR GENÉTICO/ML CONCENTRADO E SOLVENTE PARA SOLUÇÃO INJETÁVEL em Ukraine
Médicos online para LUXTURNA 5 x 10¹² UNIDADES DE VETOR GENÉTICO/ML CONCENTRADO E SOLVENTE PARA SOLUÇÃO INJETÁVEL
Avaliação de posologia, efeitos secundários, interações, contraindicações e renovação da receita de LUXTURNA 5 x 10¹² UNIDADES DE VETOR GENÉTICO/ML CONCENTRADO E SOLVENTE PARA SOLUÇÃO INJETÁVEL – sujeita a avaliação médica e regras locais.









Receba novidades da plataforma e promoções exclusivas
Fique a par das atualizações da Oladoctor e receba promoções exclusivas para subscritores.

