

LUXTURNA 5 x 10¹² GENOMAS VECTORIALES/ML CONCENTRADO Y DISOLVENTE PARA SOLUCION INYECTABLE



Cómo usar LUXTURNA 5 x 10¹² GENOMAS VECTORIALES/ML CONCENTRADO Y DISOLVENTE PARA SOLUCION INYECTABLE
Traducción generada por IA
Este contenido ha sido traducido automáticamente y se ofrece solo con fines informativos. No sustituye la consulta con un profesional sanitario.
Ver originalContenido del prospecto
Introducción
Prospecto: información para el paciente
Luxturna 5× 1012genomas vectoriales/ml concentrado y disolvente para solución inyectable
voretigén neparvovec
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de que se le administre este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o enfermero.
- Si experimenta efectos adversos, consulte a su médico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
- Qué es Luxturna y para qué se utiliza
- Qué necesita saber antes de empezar a recibir Luxturna
- Cómo se administra Luxturna
- Posibles efectos adversos
- Cómo conservar Luxturna
- Contenido del envase e información adicional
1. Qué es Luxturna y para qué se utiliza
Luxturna es un producto de terapia génica que contiene la sustancia activa voretigén neparvovec.
Luxturna se utiliza para el tratamiento de adultos y niños con pérdida de visión debido a una distrofia retiniana hereditaria causada por mutaciones en el gen RPE65.Estas mutaciones evitan que el cuerpo produzca una proteína necesaria para la visión y conducen a una pérdida de vista y a una posible ceguera.
El principio activo de Luxturna, voretigén neparvovec, es un virus modificado que contiene una copia del gen RPE65. Después de la inyección, este gen llega a las células de la retina, la capa de la parte posterior del ojo que detecta la luz. Esto permite que la retina produzca las proteínas necesarias para la visión. El virus utilizado para administrar el gen no causa enfermedad en humanos.
Únicamente se le administrará Luxturna si las pruebas genéticas muestran que su pérdida de visión es causada por mutaciones en el gen RPE65.

2. Qué necesita saber antes de empezar a recibir Luxturna
No debe recibir Luxturna
- Si es alérgico a voretigén neparvovec o a alguno de los demás componentes de este medicamento (incluidos en la sección 6)
- Si tiene una infección en el ojo
- Si tiene una inflamación en el ojo
Si usted está afectado por alguna de las situaciones anteriores, o si no está seguro, consulte a su médico antes de que se le administre Luxturna.
Advertencias y precauciones
Antes de recibir el tratamiento con Luxturna:
- Informe a su médico si tiene signos de infección ocular o inflamación ocular, por ejemplo si tiene enrojecimiento de los ojos, sensibilidad a la luz, hinchazón ocular o dolor en los ojos.
- Informe a su médico si tiene una infección activa de cualquier tipo. Su médico puede retrasar su tratamiento hasta que la infección desaparezca porque este medicamento puede dificultar la lucha contra la infección. Ver también la sección 3.
Después de recibir el tratamiento con Luxturna:
- Consulte inmediatamente a su médico si su ojo/ojos se ponen rojos, si siente dolor en los ojos, sensibilidad a la luz, ve destellos o cuerpos flotantes, o si nota un empeoramiento o visión borrosa.
- Debe evitar los viajes en avión u otros viajes a alturas elevadas hasta que su médico se lo indique. Durante el tratamiento con este medicamento, el médico inserta una burbuja de aire en el ojo, que su cuerpo absorbe lentamente. Hasta que la burbuja se haya absorbido completamente, el viaje en avión u otro viaje a alturas elevadas puede causar un crecimiento de la burbuja y provocar daños en los ojos, incluida la pérdida de visión. Consulte a su médico antes de viajar.
- Debe evitar nadar debido a un mayor riesgo de infección en el ojo. Consulte a su médico antes de ir a nadar después de recibir el tratamiento con Luxturna.
- Debe evitar la actividad física extenuante debido a un mayor riesgo de lesión en el ojo. Consulte a su médico antes de comenzar a realizar una actividad física extenuante después de recibir el tratamiento con Luxturna.
- Puede tener alteraciones visuales transitorias, como sensibilidad a la luz y visión borrosa. Informe a su médico sobre cualquier alteración visual que experimente. Su médico puede ayudarle a reducir cualquier molestia causada por estas alteraciones transitorias.
- El principio activo de Luxturna podría excretarse temporalmente por las lágrimas. Usted y su cuidador deben colocar los apósitos y el material de desecho que ha estado en contacto con lágrimas y secreciones nasales en bolsas selladas antes de desecharlos. Debe seguir estas precauciones durante 14 días.
- No podrá donar sangre, órganos, tejidos y células para el trasplante después de haber sido tratado con Luxturna.
Niños y adolescentes
Luxturna no se ha estudiado en niños menores de cuatro años de edad. Los datos son limitados.
Otros medicamentos y Luxturna
Informe a su médico si está tomando, ha tomado recientemente o pudiera tener que tomar cualquier otro medicamento.
Embarazo, lactancia y fertilidad
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o enfermero antes de recibir el tratamiento con Luxturna.
Se desconocen los efectos de este medicamento sobre el embarazo y el feto. Como precaución, no debe recibir Luxturna mientras está embarazada.
Luxturna no se ha estudiado en mujeres en periodo de lactancia. No se sabe si pasa a la leche materna. Informe a su médico si está dando el pecho o tuviera planeado hacerlo. Su médico le ayudará a decidir si tiene que interrumpir la lactancia o no recibir Luxturna, teniendo en cuenta los beneficios de la lactancia en su bebé y los beneficios de Luxturna en usted.
Conducción y uso de máquinas
Puede tener alteraciones visuales transitorias después de recibir Luxturna. No conduzca ni use máquinas pesadas hasta que su visión se haya recuperado. Consulte a su médico antes de reanudar estas actividades.
Luxturna contiene sodio
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por dosis, esto es, esencialmente "exento de sodio".
3. Cómo se administra Luxturna
Luxturna se le administrará en una sala de operaciones por cirujanos con experiencia en la realización de cirugía ocular. Luxturna se administra bajo anestesia. Su médico le hablará sobre la anestesia y sobre cómo se le administrará.
Su médico le realizará una cirugía ocular para eliminar el gel transparente que ocupa el interior del ojo, y luego le inyectará Luxturna directamente en la retina, la capa delgada sensible a la luz que se encuentra en la parte posterior del ojo. Este procedimiento se repetirá en el otro ojo al menos 6 días después. Tendrá que quedarse en observación postoperatoria durante unas horas después de cada procedimiento para controlar su recuperación y observar los efectos secundarios de la cirugía o de la anestesia.
Antes de iniciar el tratamiento con Luxturna su médico puede que le pida tomar un medicamento que suprima su sistema inmunitario (las defensas naturales del cuerpo) para que no intente luchar contra Luxturna cuando se lo administren. Es importante que tome este medicamento de acuerdo con las instrucciones que le indique su médico. No deje de tomar el medicamento sin consultar primero a su médico.
Si recibe más Luxturna del que debe
Como este medicamento se lo administrará un médico, es poco probable que se le administre más medicamento del que debe. Si esto ocurre, su médico tratará los síntomas según sea necesario. Informe a su médico o enfermero si tiene algún problema de visión.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o enfermero.

4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Pueden aparecer los siguientes efectos adversos relacionados con Luxturna:
Frecuentes (pueden afectar hasta 1de cada 10personas)
- Depósitos debajo de la retina
Frecuencia no conocida (no puede estimarse a partir de los datos disponibles)
- Atrofía de la (corio)retina
Pueden aparecer los siguientes efectos adversos relacionados con el procedimiento de inyección:
Muy frecuentes(pueden afectar a más de 1de cada 10personas)
- Enrojecimiento de los ojos
- Catarata (opacidad del cristalino)
- Aumento de la presión en el ojo
Frecuentes (pueden afectar hasta 1de cada 10personas)
- Desgarro en la retina
- Dolor en los ojos
- Hinchazón de los ojos
- Desprendimiento de la retina
- Sangrado en la parte posterior del ojo
- Dolor o aumento de las molestias en el ojo
- Visión borrosa debido a un agujero en la retina
- Adelgazamiento de la superficie del ojo (dellen)
- Irritación ocular
- Inflamación ocular
- Sensación de cuerpo extraño en el ojo
- Molestias oculares
- Anomalías en la parte posterior del ojo
- Náuseas (ganas de vomitar), vómitos, dolor abdominal (estomacal), dolor en los labios
- Cambio en la actividad eléctrica del corazón
- Dolor de cabeza, mareos
- Erupción cutánea, hinchazón de la cara
- Ansiedad
- Problemas asociados con la colocación de un tubo de respiración en la tráquea
- Rotura de la herida quirúrgica
No conocidos (la frecuencia no se puede estimar a partir de los datos disponibles)
- Enturbiamiento de la sustancia gelatinosa que se encuentra en el interior del ojo (opacidades vítreas)
- Atrofia de la (corio)retina
El daño de los tejidos del ojo puede ir acompañado de sangrado, de inflamación y de un mayor riesgo de infección. Se produce una reducción de la visión en los días posteriores a la cirugía que generalmente mejora; informe a su médico si la visión no regresa.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Luxturna
Luxturna será conservado por los profesionales sanitarios en su centro de salud.
El concentrado y el disolvente deben transportarse y almacenarse congelados a temperatura ≤ ‑65 ºC. Una vez descongelado, el medicamento no debe volver a congelarse y debe dejarse a temperatura ambiente (por debajo de 25 °C).
No utilice este medicamento después de la fecha de caducidad que aparece en la etiqueta y en la caja después de CAD.
6. Contenido del envase e información adicional
Composición de Luxturna
- El principio activo es voretigén neparvovec. Cada ml de concentrado contiene 5 × 1012 genomas vectoriales (vg). El concentrado (vial unidosis de 2 ml con volumen extraíble de 0,5 ml) requiere una dilución de 1:10 antes de la administración.
- Cada dosis de solución diluida contiene 1,5 × 1011 genomas vectoriales de voretigén neparvovec en un volumen administrable de 0,3 ml.
- Los demás excipientes del concentrado son cloruro de sodio (véase “Luxturna contiene sodio” al final de la sección 2 de este prospecto), dihidrogenofosfato de sodio monohidrato (para ajustar el pH), dihidrogenofosfato de sodio dihidrato (para ajustar el pH), poloxámero 188 y agua para preparaciones inyectables.
- El disolvente contiene cloruro de sodio (ver el final de la sección 2), dihidrogenofosfato de sodio monohidrato (para ajustar el pH), dihidrogenofosfato de sodio dihidrato (para ajustar el pH), poloxámero 188 y agua para preparaciones inyectables.
Este medicamento contiene organismos modificados genéticamente.
Aspecto de Luxturna y contenido del envase
Luxturna es un concentrado claro e incoloro para solución para inyección subretiniana que se presenta en un vial de plástico transparente. El disolvente es un líquido transparente e incoloro que se presenta en un vial de plástico transparente.
Cada bolsita de aluminio contiene una caja de cartón que incluye 1 vial de 0,5 ml de concentrado y 2 viales de disolvente (cada uno contiene 1,7 ml).
Titular de la autorización de comercialización
Novartis Europharm Limited
Vista Building
Elm Park, Merrion Road
Dublin 4
Irlanda
Responsable de la fabricación
Novartis Pharma GmbH
Roonstrasse 25
90429 Nuremberg
Alemania
Novartis Pharma GmbH
Sophie-Germain-Strasse 10
90443 Nürnberg
Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
België/Belgique/Belgien Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 | Lietuva SIA Novartis Baltics Lietuvos filialas Tel: +370 5 269 16 50 |
| Luxembourg/Luxemburg Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
Ceská republika Novartis s.r.o. Tel: +420 225 775 111 | Magyarország Novartis Hungária Kft. Tel.: +36 1 457 65 00 |
Danmark Novartis Healthcare A/S Tlf: +45 39 16 84 00 | Malta Novartis Pharma Services Inc. Tel: +356 2122 2872 |
Deutschland Novartis Pharma GmbH Tel: +49 911 273 0 | Nederland Novartis Pharma B.V. Tel: +31 88 04 52 111 |
Eesti SIA Novartis Baltics Eesti filiaal Tel: +372 66 30 810 | Norge Novartis Norge AS Tlf: +47 23 05 20 00 |
Ελλ?δα Novartis (Hellas) A.E.B.E. Τηλ: +30 210 281 17 12 | Österreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
España Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 | Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 | Portugal Novartis Farma ‑ Produtos Farmacêuticos, S.A. Tel: +351 21 000 8600 |
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 | România Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 | Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
Ísland Vistor hf. Sími: +354 535 7000 | Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 | Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
Κ?προς Novartis Pharma Services Inc. Τηλ: +357 22 690 690 | Sverige Novartis Sverige AB Tel: +46 8 732 32 00 |
Latvija SIA Novartis Baltics Tel: +371 67 887 070 |

Fecha de la última revisión de este prospecto:
Otras fuentes de información
Este prospecto está disponible en formato de archivo de audio y en un tamaño de letra grande en la página web: http://www.voretigeneneparvovec.support
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
------------------------------------------------------------------------------------------------------------------------
Esta información está destinada únicamente a profesionales sanitarios:
Precauciones que se deben tomar antes de manipular o administrar el medicamento
Este medicamento contiene organismos modificados genéticamente. Se debe usar un equipo de protección personal (que incluya bata de laboratorio, gafas de seguridad y guantes) mientras se prepara o administra voretigén neparvovec.
La presión intraocular debe controlarse adecuadamente y se debe monitorizar antes y después de la administración del medicamento.
Después de la administración, se debe indicar a los pacientes que notifiquen inmediatamente cualquier síntoma que sugiera endoftalmitis o desprendimiento de retina y deben tratarse adecuadamente.
Preparación previa a la administración
Cada envase que contiene 1 vial de concentrado y 2 viales de disolvente es para un solo uso.
Luxturna debe inspeccionarse visualmente antes de la administración. Si se detectan partículas, turbiedad o decoloración, no se debe usar el vial unidosis.
La preparación de Luxturna debe realizarse dentro de las 4 horas previas al inicio del procedimiento de administración, en condiciones asépticas y de acuerdo con el siguiente procedimiento recomendado.
Dejar descongelar a temperatura ambiente un vial de dosis única de concentrado y dos viales de disolvente. Una vez descongelados los 3 viales (1 vial de concentrado y 2 viales de disolvente), debe iniciarse la dilución. Invertir suavemente los viales cinco veces para mezclar los contenidos.
Inspeccionar si hay partículas visibles o cualquier anomalía. La aparición de cualquier anomalía o de partículas visibles debe ser notificada al Titular de la Autorización de Comercialización y el producto no debe ser utilizado.
Transferir 2,7 ml de disolvente procedente de los dos viales descongelados y dispensar con una jeringa de 3 ml en un vial estéril de vidrio vacío de 10 ml.
Para la dilución, extraer 0,3 ml de concentrado descongelado con una jeringa de 1 ml y agregar al vial estéril de 10 ml que contiene el disolvente. Invertir suavemente el vial al menos cinco veces para una mezcla adecuada. Inspeccionar si hay partículas visibles. La solución diluida debe ser clara o ligeramente opalescente. Etiquetar el vial de vidrio de 10 ml que contiene el concentrado diluido de la siguiente manera: "Luxturna diluido".
No se deben preparar las jeringas si el vial muestra algún daño o si se observan partículas visibles. Preparar las jeringas para inyección extrayendo 0,8 ml de la solución diluida en una jeringa estéril de 1 ml. Repetir el mismo procedimiento para preparar una jeringa de repuesto. Las jeringas llenas de producto deben transportarse hasta el quirófano en un contenedor asignado para éste fin.
Medidas que deben adoptarse en caso de exposición accidental
Se debe evitar la exposición accidental. Debe seguirse la normativa local sobre bioseguridad para la preparación, administración y manejo de voretigén neparvovec.
- Se debe usar un equipo de protección personal (que incluya bata de laboratorio, gafas de seguridad y guantes) mientras se manipula o administra voretigén neparvovec.
- Debe evitarse la exposición accidental a voretigén neparvovec, incluido el contacto con la piel, los ojos y las membranas mucosas. Debe cubrirse cualquier herida antes de manipular este medicamento.
- Cualquier derramamiento de voretigén neparvovec se debe tratar con un agente virucida, como hipoclorito de sodio al 1 %, y se debe secar con materiales absorbentes.
- Todos los materiales que puedan haber entrado en contacto con voretigén neparvovec (p. ej., vial, jeringa, aguja, gasas de algodón, guantes, máscaras o vendajes) deben eliminarse de acuerdo con la normativa local sobre bioseguridad.
Exposición accidental
- En caso de exposición ocupacional accidental (p. ej., por salpicaduras en los ojos o en las membranas mucosas), se debe enjuagar con agua limpia durante al menos 5 minutos.
- En caso de exposición en la piel lesionada o de lesión por punción con la aguja, se debe limpiar bien el área afectada con agua y jabón y /o con un desinfectante.
Precauciones que se deben tomar en la eliminación del medicamento
Este medicamento contiene organismos modificados genéticamente. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local de residuos farmacéuticos.
Posología
El tratamiento debe ser iniciado y administrado por un cirujano de retina con experiencia en cirugía macular.
Los pacientes recibirán una dosis única de voretigén neparvovec de 1,5 × 1011 genomas vectoriales en cada ojo. Cada dosis se administrará dentro del espacio subretiniano en un volumen total de 0,3 ml. La administración debe realizarse de forma individualizada en cada ojo en días separados dentro de un corto intervalo de, al menos, seis días de diferencia entre cada procedimiento quirúrgico.
Pauta inmunomoduladora
Antes de iniciar la pauta inmunomoduladora y antes de la administración de voretigén neparvovec, debe examinarse al paciente para detectar síntomas de enfermedad infecciosa activa de cualquier naturaleza, y en caso de tal infección, el inicio del tratamiento debe posponerse hasta después de que el paciente se haya recuperado.
Se recomienda iniciar la pauta inmunomoduladora 3 días antes de la administración de voretigén neparvovec en el primer ojo, siguiendo el calendario descrito a continuación (Tabla 1). El inicio de la pauta inmunomoduladora para el segundo ojo debe seguir el mismo esquema y debe reemplazar a la pauta inmunomoduladora del primer ojo.
Tabla1Pauta inmunomoduladora pre y postoperatoria para cada ojo
Preoperatorio | 3 días antes de la administración de Luxturna | Prednisona (o equivalente) 1 mg/kg/día (hasta un máximo de 40 mg/día) |
Postoperatorio | 4 días (incluyendo el día de la administración) | Prednisona (o equivalente) 1 mg/kg/día (hasta un máximo de 40 mg/día) |
Continuar 5 días | Prednisona (o equivalente) 0,5 mg/kg/día (hasta un máximo de 20 mg/día) | |
Continuar 5 días con una dosis cada dos días | Prednisona (o equivalente) 0,5 mg/kg cada dos días (hasta un máximo de 20 mg/día) |
Poblaciones especiales
Pacientes de edad avanzada
No se ha establecido la seguridad y eficacia de voretigén neparvovec en pacientes ≥ 65 años. Los datos son limitados. Sin embargo, no es necesario un ajuste de la dosis en pacientes de edad avanzada.
Insuficiencia hepática y renal
No se ha establecido la seguridad y eficacia de voretigén neparvovec en pacientes con insuficiencia hepática o renal. No es necesario un ajuste de la dosis en estos pacientes (ver sección 5.2).
Población pediátrica
No se ha establecido la seguridad y eficacia de voretigén neparvovec en niños menores de 4 años de edad. Los datos son limitados. No es necesario un ajuste de la dosis en pacientes pediátricos.
Forma de administración
Uso subretinal.
Luxturna es una solución estéril concentrada para inyección subretiniana que requiere descongelación y dilución antes de la administración.
Este medicamento no debe administrarse mediante inyección intravítrea.
Luxturna es un vial de un solo uso para una administración única en un solo ojo. El producto se administra mediante una inyección subretiniana tras realizar una vitrectomía en cada ojo. No se debe administrar muy próximo a la fóvea para mantener la integridad foveal.
La administración de voretigén neparvovec debe llevarse a cabo en el quirófano bajo unas condiciones asépticas controladas. Antes del procedimiento, se debe administrar al paciente la anestesia adecuada. La pupila del ojo en el que se va a administrar la inyección debe estar dilatada, y antes de la cirugía se debe administrar un antibiótico de amplio espectro por vía oftálmica de acuerdo con la práctica médica habitual.
Administración
Seguir los pasos descritos a continuación para administrar voretigén neparvovec a los pacientes:
- Una vez diluido Luxturna, debe inspeccionarse visualmente antes de la administración. Si se observan partículas, turbiedad o decoloración, el medicamento no debe utilizarse.
- Conectar la jeringa que contiene el producto diluido al tubo de extensión y la cánula de inyección subretiniana. El producto se debe inyectar lentamente a través del tubo de extensión y la cánula de inyección subretiniana para eliminar cualquier burbuja de aire en el sistema.
- El volumen de producto disponible para inyección se confirma en la jeringa al alinear la punta del émbolo con la línea que marca 0,3 ml.
- Una vez finalizada la vitrectomía, Luxturna se administra mediante inyección subretiniana utilizando una cánula de inyección subretiniana introducida por vía pars plana.
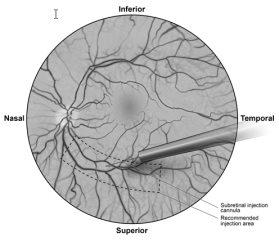
- Bajo visualización directa, la punta de la cánula de la inyección subretiniana se pone en contacto con la superficie de la retina. El sitio de inyección recomendado debería situarse a lo largo de la arcada vascular superior, al menos a 2 mm de distancia del centro de la fóvea. Se inyecta lentamente una pequeña cantidad de producto hasta que se observa una ampolla subretiniana inicial, y luego el volumen restante se inyecta lentamente hasta que se administran los 0,3 ml totales (Figura 1).
Figura1Punta de la cánula de inyección subretiniana colocada en el sitio de inyección recomendado (vista del cirujano)
- Al finalizar la inyección, se retira la cánula de inyección subretiniana del ojo.
- Después de la inyección, se debe desechar cualquier producto no utilizado. No se debe guardar la jeringa de repuesto.
- Se debe realizar cuidadosamente un intercambio fluido-aire, evitando el drenaje de líquido cerca de la retinotomía creada para la inyección subretiniana.
- En el postoperatorio se debe colocar la cabeza en posición supina inmediatamente y se debe mantener durante 24 horas tras el alta del paciente.
- País de registro
- Principio activo
- Requiere recetaSí
- Fabricante
- Esta información es de carácter general y no sustituye la consulta con un profesional sanitario.
- Alternativas a LUXTURNA 5 x 10¹² GENOMAS VECTORIALES/ML CONCENTRADO Y DISOLVENTE PARA SOLUCION INYECTABLEForma farmacéutica: COLIRIO, 5,5 mg sodio cloruro;3 mg hipomelosa/mlPrincipio activo: artificial tears and other indifferent preparationsFabricante: Alcon Healthcare S.A.No requiere recetaForma farmacéutica: COLIRIO, 3,2 mg/mlPrincipio activo: artificial tears and other indifferent preparationsFabricante: Bausch & Lomb S.A.No requiere recetaForma farmacéutica: COLIRIO, 3,2 mg/mlPrincipio activo: artificial tears and other indifferent preparationsFabricante: Bausch & Lomb S.A.No requiere receta
Médicos online para LUXTURNA 5 x 10¹² GENOMAS VECTORIALES/ML CONCENTRADO Y DISOLVENTE PARA SOLUCION INYECTABLE
Comenta la dosis, los posibles efectos secundarios, interacciones, contraindicaciones o la revisión de receta de LUXTURNA 5 x 10¹² GENOMAS VECTORIALES/ML CONCENTRADO Y DISOLVENTE PARA SOLUCION INYECTABLE, sujeto a valoración médica y a la normativa local.









Preguntas frecuentes
