

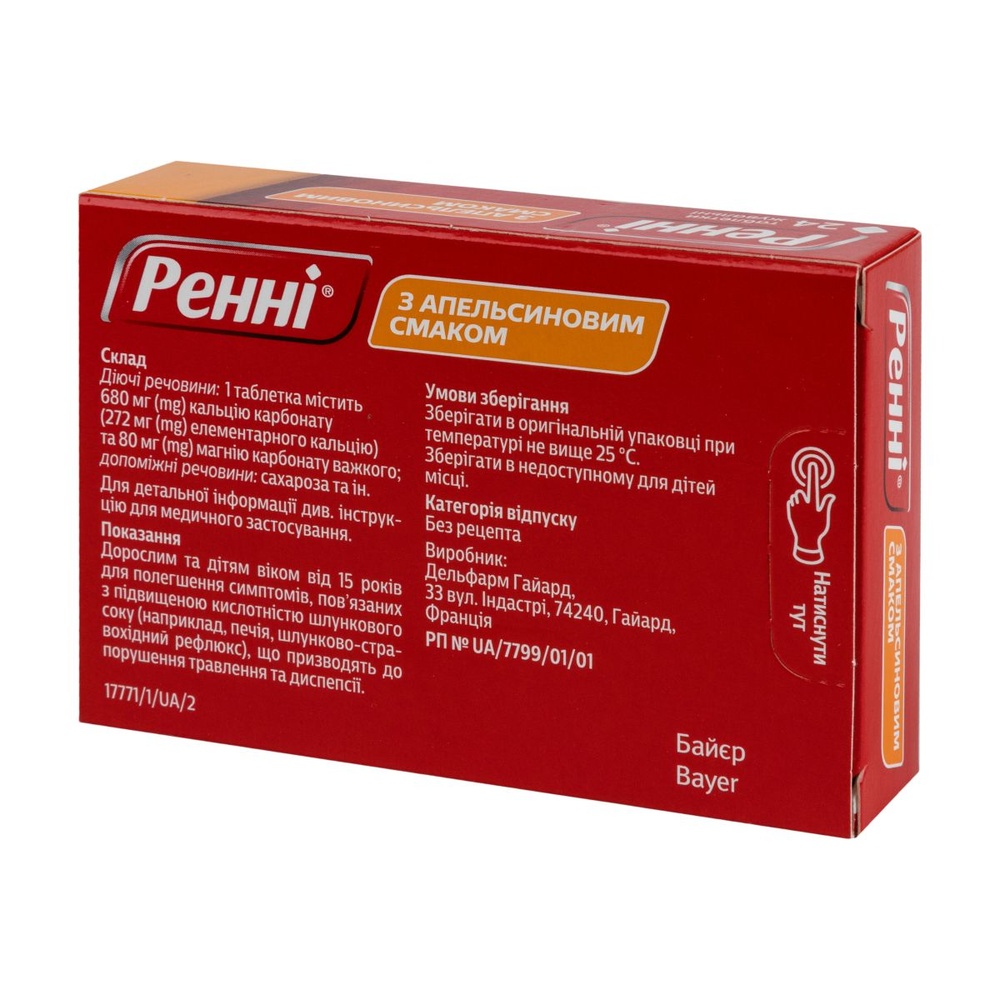
RENNI Z APEL'SINOVIM SMAKOM
Consulta con un médico sobre la receta médica de RENNI Z APEL'SINOVIM SMAKOM



Cómo usar RENNI Z APEL'SINOVIM SMAKOM
INSTRUCCIONES para la aplicación médica del medicamento JAKAVI (JAKAVI®)
COMPOSICIÓN
Principio activo: ruxolitinib; 1 tableta contiene 5 mg, 15 mg o 20 mg de ruxolitinib (en forma de fosfato); excipientes: monohidrato de lactosa, celulosa microcristalina, glicolato de almidón de sodio (tipo A), hidroxipropilcelulosa, povidona, dióxido de silicio coloidal anhidro, estearato de magnesio.
FORMA FARMACÉUTICA
Tabletas.
PROPIEDADES FÍSICO-QUÍMICAS FUNDAMENTALES
Tabletas de 5 mg: redondas, abombadas, de color blanco a casi blanco, de aproximadamente 7,5 mm de diámetro, con la inscripción "NVR" en un lado y "L5" en el otro; tabletas de 15 mg: ovaladas, abombadas, de color blanco a casi blanco, de aproximadamente 15,0 x 7,0 mm, con la inscripción "NVR" en un lado y "L15" en el otro; tabletas de 20 mg: alargadas, abombadas, de color blanco a casi blanco, de aproximadamente 16,5 x 7,4 mm, con la inscripción "NVR" en un lado y "L20" en el otro.
GRUPO FARMACOTERAPÉUTICO
Agentes antineoplásicos. Inhibidores de proteínas quinasas. Inhibidores de la quinasa Janus (JAK).
CÓDIGO ATC
L01E J01.
PROPIEDADES FARMACOLÓGICAS
FARMACODINAMIA
El ruxolitinib es un inhibidor selectivo de las quinasas Janus (JAK) JAK1 y JAK2 (con valores de IC50 para las enzimas JAK1 y JAK2 de 3,3 nM y 2,8 nM, respectivamente). Estas enzimas son intermediarias en la transmisión de señales de varias citocinas y factores de crecimiento que son importantes para la hematopoyesis y el sistema inmunológico. La mielofibrosis (MF) y la policitemia vera (PV) son neoplasias mieloproliferativas que, según se sabe, están asociadas con una alteración de la regulación en la transmisión de señales que involucran a JAK1 y JAK2. La base de la alteración de la regulación es la presencia de altos niveles de citocinas circulantes que activan la vía de señalización JAK-STAT-mutaciones, en las que el producto de la expresión del gen mutante adquiere nuevas funciones patológicas, como la mutación JAK2V617F, así como también conduce a la exclusión de mecanismos reguladores negativos. En pacientes con MF, se observa una alteración de la regulación en la transmisión de señales que involucran a JAK, independientemente del estado de mutación JAK2V617F. Las mutaciones activadoras de JAK2 (V617F o exón 12) se observan en más del 95% de los pacientes con PV. El ruxolitinib inhibe la transmisión de señales de la vía JAK-STAT y la proliferación de células en modelos celulares de neoplasias hematológicas dependientes de citocinas, así como en células Ba/F3 que expresan la proteína JAK2V617F modificada, con un IC50 que se encuentra en el rango de 80-320 nM.
EFECTOS FARMACODINÁMICOS
El ruxolitinib inhibe la fosforilación de STAT3 inducida por citocinas en sangre total de voluntarios sanos y pacientes con MF y PV. La administración de ruxolitinib conduce a una inhibición máxima de la fosforilación de STAT3 dentro de las 2 horas después de la administración del medicamento, con un regreso a los niveles de fosforilación cercanos a los niveles iniciales dentro de las 8 horas, tanto en voluntarios sanos como en pacientes con MF, lo que indica la ausencia de acumulación del medicamento original o de metabolitos activos. Los niveles elevados de marcadores de inflamación asociados con la aparición de síntomas sistémicos, como el factor de necrosis tumoral alfa (FNT-α), la interleucina-6 (IL-6) y la proteína C reactiva (PCR), en pacientes con MF, disminuyeron después del tratamiento con ruxolitinib. Con el tiempo, en pacientes con MF, no se desarrollaron signos de refractariedad a los efectos farmacodinámicos del tratamiento con ruxolitinib. De manera similar, en pacientes con PV, se observó un aumento en los niveles iniciales de marcadores de inflamación, y estos niveles disminuyeron después del tratamiento con ruxolitinib. En un estudio detallado de la duración del intervalo QT en voluntarios sanos, no se encontraron signos de prolongación del QT/QTc con la administración de ruxolitinib en dosis únicas que excedían las dosis terapéuticas (hasta 200 mg), lo que sugiere que el ruxolitinib no tiene efecto sobre la repolarización cardíaca.
FARMACOCINÉTICA
ABSORCIÓN
El ruxolitinib pertenece a las sustancias de la clase 1 según el Sistema de clasificación biofarmacéutica (BCS) con alta permeabilidad, alta solubilidad y disolución rápida. En el curso de los estudios clínicos, se determinó que el ruxolitinib se absorbe rápidamente después de la administración oral, alcanzando la concentración máxima del medicamento en plasma sanguíneo (Cmax) aproximadamente 1 hora después de la administración de la dosis. Teniendo en cuenta los datos del estudio de balance de masa corporal en humanos, la absorción del ruxolitinib después de la administración oral del medicamento en forma de ruxolitinib o metabolitos formados durante el primer paso es del 95% o más. La Cmax media y la exposición total (AUC) del ruxolitinib aumentaron proporcionalmente con el aumento de la dosis única en el rango de 5 a 200 mg. No se observaron cambios clínicamente significativos en la farmacocinética del ruxolitinib después de la administración del medicamento con alimentos con alto contenido de grasa. Durante la administración del medicamento con alimentos con alto contenido de grasa, la Cmax media se redujo moderadamente (un 24%), mientras que el valor medio de AUC permaneció casi sin cambios (aumento del 4%).
DISTRIBUCIÓN
En pacientes con MF y PV, el volumen medio de distribución en estado de equilibrio es de aproximadamente 75 litros. Las concentraciones clínicamente significativas de ruxolitinib en la unión a proteínas plasmáticas (principalmente albúmina) in vitro son de aproximadamente el 97%. Un estudio de autorradiografía de todo el organismo en ratas mostró que el ruxolitinib no cruza la barrera hematoencefálica.
BIOTRANSFORMACIÓN
El ruxolitinib se metaboliza principalmente por CYP3A4 (>50%) con participación adicional de la enzima CYP2C9. La sustancia original es la forma dominante en plasma sanguíneo humano, representando alrededor del 60% de la sustancia relacionada con el medicamento en la circulación. En plasma sanguíneo, se detectaron dos metabolitos principales y activos, que representan el 25% y el 11% de la AUC de la sustancia original, respectivamente. Estos metabolitos tienen de 1/2 a 1/5 de la actividad farmacológica relacionada con JAK de la sustancia original. En conjunto, la suma de todos los metabolitos activos proporciona hasta el 18% de la farmacodinamia total del ruxolitinib. En concentraciones clínicamente significativas, el ruxolitinib no inhibe la actividad de las enzimas CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 o CYP3A4, y no es un inhibidor potente de las enzimas CYP1A2, CYP2B6 o CYP3A4, según los resultados de los estudios in vitro. Los datos obtenidos in vitro indican que el ruxolitinib puede inhibir la actividad de la glicoproteína P y la proteína de resistencia a la lactancia (BCRP).
ELIMINACIÓN
El ruxolitinib se elimina principalmente por metabolismo. El período medio de eliminación del ruxolitinib es de aproximadamente 3 horas. Después de la administración de una dosis única de [14C]-ruxolitinib marcado en voluntarios sanos adultos, la eliminación se produjo principalmente por metabolismo, con el 74% de la sustancia radiactivamente marcada eliminada en la orina y el 22% en las heces. La sustancia original no modificada representó menos del 1% de la sustancia radiactivamente marcada eliminada.
LINEALIDAD/NO LINEALIDAD
La proporcionalidad de la dosis se demostró en los estudios de administración de dosis única y múltiple del medicamento.
GRUPOS ESPECIALES DE PACIENTES
EFECTO DE LA SUPERFICIE CORPORAL, LA EDAD, EL SEXO O LA RAZA
En los estudios con voluntarios sanos de diferentes sexos y razas, no se observaron diferencias significativas en la farmacocinética del ruxolitinib. Durante la evaluación farmacocinética poblacional en pacientes con MF, no se encontró ninguna dependencia aparente de la eliminación oral con respecto a la edad o la raza del paciente. El aclaramiento oral pronóstico en pacientes con MF fue de 17,7 l/h en mujeres y 22,1 l/h en hombres, con una variabilidad del 39%. En pacientes con PV, el aclaramiento fue de 12,7 l/h, con una variabilidad del 42%. Durante la evaluación farmacocinética poblacional en pacientes con PV, no se encontró ninguna dependencia del aclaramiento oral con respecto al sexo, la edad o la raza del paciente. El aclaramiento fue de 10,4 l/h en pacientes con TTP aguda y 7,8 l/h en pacientes con TTP crónica, con una variabilidad interindividual del 49%. Durante la evaluación farmacocinética poblacional en pacientes con TTP, no se encontró ninguna dependencia del aclaramiento oral con respecto al sexo, la edad o la raza del paciente. La exposición aumentó en pacientes con TTP y baja superficie corporal. En pacientes con una superficie corporal de 1 m2, 1,25 m2 y 1,5 m2, la exposición media pronóstica (AUC) fue un 31%, 22% y 12% más alta, respectivamente, que en una persona adulta promedio (1,79 m2).
POBLACIÓN PEDIÁTRICA
La farmacocinética del medicamento JAKAVI en niños (menores de 18 años) con MF y PV no se ha establecido. El perfil farmacocinético observado en niños con TTP aguda o crónica fue comparable al de la población general de pacientes.
El ruxolitinib no ha sido estudiado en niños menores de 12 años con TTP aguda o crónica.
DISFUNCIÓN RENAL
La función renal se evaluó utilizando la dieta modificada para la enfermedad renal (MDRD) y el aclaramiento de creatinina. Después de la administración de una dosis única de 25 mg de ruxolitinib, su exposición fue similar en pacientes con diferentes grados de disfunción renal y en personas con función renal normal. Sin embargo, los valores de AUC de los metabolitos de ruxolitinib en plasma sanguíneo aumentaron con la gravedad de la disfunción renal y fueron más pronunciados en pacientes con disfunción renal grave. No se sabe si el aumento de la exposición a los metabolitos tiene implicaciones para la seguridad. Se recomienda ajustar la dosis del medicamento para pacientes con disfunción renal grave y enfermedad renal terminal (ver sección "Modo de administración y dosis"). La administración del medicamento solo en los días de diálisis reduce la exposición a los metabolitos y el efecto farmacodinámico, especialmente en los días entre las sesiones de diálisis.
DISFUNCIÓN HEPÁTICA
Después de la administración de una dosis única de 25 mg de ruxolitinib, el valor medio de AUC de ruxolitinib aumentó en pacientes con disfunción hepática leve, moderada y grave en un 87%, 28% y 65%, respectivamente, en comparación con pacientes con función hepática normal. No se encontró una relación clara entre el AUC del medicamento y el grado de disfunción hepática según la puntuación de la escala de Child-Pugh. El período terminal de eliminación se prolongó en pacientes con disfunción hepática en comparación con el grupo control de voluntarios sanos (4,1-5 horas frente a 2,8 horas). Se recomienda reducir la dosis del medicamento aproximadamente en un 50% para pacientes con MF y PV que tienen disfunción hepática (ver sección "Modo de administración y dosis").
Para pacientes con TTP y disfunción hepática no relacionada con TTP, se debe reducir la dosis inicial de ruxolitinib en un 50%.
DATOS PRECLÍNICOS DE SEGURIDAD
El ruxolitinib se evaluó en estudios farmacológicos de seguridad, estudios de toxicidad de dosis repetidas, genotoxicidad y toxicidad reproductiva, así como en estudios de carcinogenicidad. Los órganos diana relacionados con el efecto farmacológico del ruxolitinib en los estudios de dosis repetidas incluyeron la médula ósea, la sangre periférica y los tejidos linfoides. Las infecciones, comúnmente asociadas con la inmunosupresión, se observaron en perros. La disminución de la presión arterial y el aumento de la frecuencia cardíaca se observaron en un estudio en perros utilizando mediciones telemétricas; la disminución del volumen minuto respiratorio se observó en un estudio de la función del tracto respiratorio en ratas. El estudio de autorradiografía de todo el organismo en ratas mostró que el ruxolitinib no cruza la barrera hematoencefálica.
CARACTERÍSTICAS CLÍNICAS
INDICACIONES
Mielofibrosis. Tratamiento de enfermedades asociadas con esplenomegalia o síntomas de mielofibrosis primaria (también conocida como mielofibrosis idiopática crónica) en pacientes adultos, mielofibrosis secundaria a policitemia vera o mielofibrosis secundaria a trombocitemia esencial.
Policitemia vera. Tratamiento de la policitemia vera en pacientes adultos con resistencia o intolerancia a la hidroxiurea.
Reacción de "trasplante contra huésped" (TTPH). Tratamiento de pacientes de 12 años de edad o más con reacción aguda de TTPH o reacción crónica de TTPH que tienen una respuesta inadecuada al tratamiento con corticosteroides o otros medicamentos sistémicos.
CONTRAINDICACIONES
Hipersensibilidad aumentada al principio activo o a alguno de los excipientes del medicamento. Embarazo y lactancia.
INTERACCIONES CON OTROS MEDICAMENTOS Y OTROS TIPOS DE INTERACCIONES
Los estudios de interacción se realizaron solo con participación de adultos.
El ruxolitinib se elimina del organismo mediante metabolismo, que es catalizado por CYP3A4 y CYP2C9. Por lo tanto, la administración de medicamentos que inhiben la actividad de estas enzimas puede conducir a un aumento de la exposición al ruxolitinib.
INTERACCIONES QUE REQUIEREN UNA REDUCCIÓN DE LA DOSIS DE RUXOLITINIB
Inhibidores de CYP3A4
Inhibidores potentes de CYP3A4 (boceprevir, claritromicina, indinavir, itraconazol, ketconazol, lopinavir/ritonavir, mibefradil, nefazodona, nelfinavir, posaconazol, saquinavir, telaprevir, telitromicina, voriconazol, etc.)
En voluntarios sanos, la administración concomitante de ruxolitinib (dosis única de 10 mg) con un inhibidor potente de CYP3A4, ketconazol, condujo a un aumento de los valores de Cmax y AUC de ruxolitinib en un 33% y un 91%, respectivamente, en comparación con los valores de Cmax y AUC cuando se administró ruxolitinib solo. El período de eliminación se prolongó de 3,7 a 6 horas cuando se administró ruxolitinib con ketconazol.
Al administrar ruxolitinib con inhibidores potentes de CYP3A4, se debe reducir la dosis estándar de ruxolitinib aproximadamente en un 50% y administrarla dos veces al día. Los pacientes deben ser cuidadosamente monitoreados (por ejemplo, dos veces a la semana) para el desarrollo de citopenia, y el titrado de la dosis del medicamento debe realizarse con respecto a la seguridad y la eficacia (ver sección "Modo de administración y dosis").
Inhibidores dobles de CYP2C9 y CYP3A4
En voluntarios sanos, la administración concomitante de ruxolitinib (dosis única de 10 mg) con un inhibidor doble de CYP2C9 y CYP3A4, fluconazol, condujo a un aumento de los valores de Cmax y AUC de ruxolitinib en un 47% y un 232%, respectivamente, en comparación con los valores de Cmax y AUC cuando se administró ruxolitinib solo.
Se debe considerar la reducción de la dosis del medicamento en un 50% cuando se administra con medicamentos que son inhibidores dobles de CYP2C9 y CYP3A4 (por ejemplo, fluconazol). Se debe evitar la administración de ruxolitinib con fluconazol en dosis que excedan los 200 mg al día.
OTRAS INTERACCIONES QUE AFECTAN AL RUXOLITINIB
Inhibidores leves o moderados de CYP3A4 (ciprofloxacino, eritromicina, amprenavir, atazanavir, diltiazem, cimetidina, etc.)
En voluntarios sanos, la administración concomitante de ruxolitinib (dosis única de 10 mg) con eritromicina a una dosis de 500 mg dos veces al día durante cuatro días condujo a un aumento de los valores de Cmax y AUC de ruxolitinib en un 8% y un 27%, respectivamente, en comparación con los valores de Cmax y AUC cuando se administró ruxolitinib solo.
Al administrar ruxolitinib con inhibidores leves o moderados de CYP3A4 (por ejemplo, eritromicina), no se recomienda la corrección de la dosis de ruxolitinib. Sin embargo, al inicio del tratamiento con ruxolitinib junto con un inhibidor moderado de CYP3A4, los pacientes deben estar bajo vigilancia cuidadosa para la aparición de citopenia.
EFECTO DEL RUXOLITINIB EN OTROS MEDICAMENTOS
Sustancias transportadas por la glicoproteína P o otros transportadores
El ruxolitinib puede inhibir la actividad de la glicoproteína P y la proteína de resistencia a la lactancia (BCRP) en el intestino. Esto puede conducir a un aumento de la exposición sistémica de los sustratos de estos transportadores, como el etexilato de dabigatrán, la ciclosporina, el rosuvastatina y, posiblemente, la digoxina. Se recomienda el monitoreo terapéutico o clínico de los sustratos involucrados.
Es posible que la inhibición esperada de la glicoproteína P y la proteína de resistencia a la lactancia en el intestino se minimice si el intervalo de tiempo entre la administración de los medicamentos es lo más largo posible.
Un estudio con voluntarios sanos demostró que el ruxolitinib no inhibió el metabolismo de la midazolam, que se administra por vía oral y es un sustrato de CYP3A4. Por lo tanto, no se espera un aumento de la exposición a los sustratos de CYP3A4 cuando se administra concomitantemente con ruxolitinib. En otro estudio con voluntarios sanos, se determinó que el ruxolitinib no afectó la farmacocinética de los anticonceptivos orales que contienen etinilestradiol y levonorgestrel. Por lo tanto, no se espera que el ruxolitinib afecte la eficacia de los anticonceptivos orales cuando se administra concomitantemente.
CARACTERÍSTICAS ESPECIALES
MIELOSUPRESIÓN
El tratamiento con el medicamento JAKAVI puede conducir a la aparición de reacciones adversas hematológicas, incluyendo trombocitopenia, anemia y neutropenia. Antes de iniciar el tratamiento con JAKAVI, se debe realizar un análisis de sangre completo, incluyendo un recuento diferencial de leucocitos. El tratamiento debe suspenderse en pacientes con un recuento de plaquetas inferior a 50.000/μL (50 x 10^9/L) o un recuento absoluto de neutrófilos inferior a 500/μL (0,5 x 10^9/L) (ver sección "Modo de administración y dosis").
Se ha determinado que los pacientes con MF y un bajo nivel de plaquetas (<200.000/μL (<200 x 10^9/L)) en el inicio del tratamiento son más propensos a desarrollar trombocitopenia durante el tratamiento.
La trombocitopenia, en general, es reversible y, por lo general, se corrige mediante la reducción de la dosis del medicamento o la suspensión temporal del tratamiento con JAKAVI (ver secciones "Modo de administración y dosis" y "Reacciones adversas"). Sin embargo, según las indicaciones clínicas, puede ser necesario la transfusión de plaquetas.
Los pacientes con anemia pueden requerir transfusión de sangre. Para los pacientes que desarrollan anemia, es posible cambiar la dosis del medicamento o suspender el tratamiento.
Los pacientes con un nivel de hemoglobina inferior a 10,0 g/dL (100 g/L) en el inicio del tratamiento tienen un mayor riesgo de disminución del nivel de hemoglobina por debajo de 8 g/dL (80 g/L) durante el tratamiento en comparación con los pacientes con un nivel de hemoglobina más alto en el inicio del tratamiento (79,3% en comparación con 30,1%). Para los pacientes con un nivel de hemoglobina inferior a 10,0 g/dL (100 g/L) en el inicio del tratamiento, se recomienda un control más frecuente de los parámetros hematológicos y los síntomas clínicos de las reacciones adversas relacionadas con el uso de JAKAVI. La neutropenia (recuento absoluto de neutrófilos <500/μL (<0,5 x 10^9/L)), en general, fue reversible y, por lo general, se corrigió mediante la suspensión temporal del tratamiento con JAKAVI (ver secciones "Modo de administración y dosis" y "Reacciones adversas"). Según las indicaciones clínicas, es necesario realizar un análisis de sangre completo y ajustar las dosis del medicamento según sea necesario (ver secciones "Modo de administración y dosis" y "Reacciones adversas").
INFECCIONES
En pacientes que tomaron el medicamento JAKAVI, se observaron infecciones bacterianas graves, micobacterianas, fúngicas, virales y otras infecciones oportunistas. El estado de los pacientes debe evaluarse en cuanto al riesgo de desarrollar infecciones graves. Los médicos deben vigilar cuidadosamente a los pacientes que toman JAKAVI para detectar síntomas de infecciones y iniciar el tratamiento adecuado si es necesario. No se debe iniciar el tratamiento con JAKAVI mientras no se hayan resuelto las infecciones activas graves.
En pacientes que tomaron JAKAVI, se informaron casos de tuberculosis. Según las recomendaciones locales, antes de iniciar el tratamiento, los pacientes deben ser examinados para detectar tuberculosis activa o latente ("oculta"), lo que puede incluir el estudio de la historia clínica, la determinación de la posibilidad de contacto previo con pacientes con tuberculosis y/o la realización de pruebas de detección adecuadas, como radiografías de tórax, pruebas de tuberculina y/o análisis de liberación de interferón gamma, según las circunstancias. Existe el riesgo de resultados falsos negativos de la prueba de tuberculina cutánea, especialmente en pacientes con enfermedades graves o inmunidad debilitada.
En pacientes con hepatitis viral crónica B que tomaron JAKAVI, se observaron aumentos de la carga viral (título de ADN de VHB) con aumentos de los niveles de alanina aminotransferasa y aspartato aminotransferasa, o sin ellos. Antes de iniciar el tratamiento con JAKAVI, se recomienda realizar pruebas para detectar la infección por VHB. Los pacientes con infección crónica por VHB deben recibir tratamiento y seguimiento según las recomendaciones clínicas.
HERPES ZÓSTER
Los médicos deben informar a los pacientes sobre la aparición de síntomas tempranos de herpes zóster, informándoles de que el tratamiento debe iniciarse lo antes posible.
LEUCOENCEFALOPATÍA MULTIFOCAL PROGRESIVA
En pacientes que tomaron JAKAVI, se informaron casos de leucoencefalopatía multifocal progresiva (LMP). Los médicos deben ser especialmente cuidadosos con los síntomas que pueden indicar LMP, que los pacientes pueden no notar (por ejemplo, síntomas cognitivos, neurológicos o psiquiátricos). Los pacientes deben estar bajo control para detectar la aparición de nuevos síntomas o el empeoramiento de los síntomas existentes, y si estos síntomas aparecen, se debe considerar la necesidad de consultar a un neurólogo y realizar pruebas de detección adecuadas para detectar LMP. Si se sospecha LMP, el tratamiento con JAKAVI debe suspenderse hasta que se descarte la sospecha de LMP.
ALTERACIONES DEL METABOLISMO DE LOS LÍPIDOS
El tratamiento con JAKAVI se ha asociado con aumentos de los niveles de lípidos, incluyendo colesterol total, colesterol de lipoproteínas de alta densidad (LDL), colesterol de lipoproteínas de baja densidad (LDL) y triglicéridos. Se recomienda el control de los niveles de lípidos y el tratamiento de la dislipidemia según las recomendaciones clínicas.
REACCIONES ADVERSAS CARDÍACAS PRINCIPALES (MACE)
En un estudio aleatorizado grande con control activo de tofacitinib (otro inhibidor de JAK) en pacientes de 50 años de edad o más con artritis reumatoide y al menos un factor de riesgo adicional de enfermedad cardiovascular, se observó una frecuencia más alta de MACE, definidos como muerte cardiovascular, infarto de miocardio no mortal (IM) y accidente cerebrovascular no mortal, con tofacitinib en comparación con los inhibidores del factor de necrosis tumoral (FNT).
Se han informado casos de MACE en pacientes que recibieron JAKAVI. Antes de iniciar o continuar el tratamiento con JAKAVI, se debe sopesar el beneficio y el riesgo para cada paciente individual, especialmente para los pacientes de 65 años de edad o más, los pacientes que fuman o han fumado durante un período prolongado, y los pacientes con enfermedad cardiovascular aterosclerótica en su historial o otros factores de riesgo de enfermedad cardiovascular en su historial.
TROMBOSIS
En un estudio aleatorizado grande con control activo de tofacitinib (otro inhibidor de JAK) en pacientes de 50 años de edad o más con artritis reumatoide y al menos un factor de riesgo adicional de enfermedad cardiovascular, se observó una frecuencia más alta de tromboembolia venosa (TEV), incluyendo trombosis de venas profundas (TVP) y tromboembolia pulmonar (TEP), con tofacitinib en comparación con los inhibidores del factor de necrosis tumoral (FNT).
En pacientes que recibieron JAKAVI, se informaron casos de trombosis de venas profundas (TVP) y tromboembolia pulmonar (TEP). En los estudios clínicos con pacientes con mielofibrosis y policitemia vera, la frecuencia de tromboembolia fue comparable en el grupo de tratamiento con JAKAVI y el grupo control.
Antes de iniciar o continuar el tratamiento con JAKAVI, se debe sopesar el beneficio y el riesgo para cada paciente individual, especialmente para los pacientes con factores de riesgo de enfermedad cardiovascular (ver también "Reacciones adversas cardíacas principales (MACE)").
Los pacientes con síntomas de trombosis deben ser examinados de inmediato y recibir el tratamiento adecuado.
NEOPLASIAS SECUNDARIAS, DETECTADAS POR PRIMERA VEZ
En un estudio aleatorizado grande con control activo de tofacitinib (otro inhibidor de JAK) en pacientes de 50 años de edad o más con artritis reumatoide y al menos un factor de riesgo adicional de enfermedad cardiovascular, se observó una frecuencia más alta de neoplasias malignas, en particular cáncer de pulmón, linfomas y cáncer de piel no melanoma (CPNM), con tofacitinib en comparación con los inhibidores del factor de necrosis tumoral (FNT).
En pacientes que reciben inhibidores de JAK, incluyendo JAKAVI, se han informado casos de linfomas y otras neoplasias malignas.
El cáncer de piel no melanoma (CPNM), incluyendo cáncer de células basales, cáncer de células escamosas y carcinoma de células de Merkel, se observó en pacientes que recibieron ruxolitinib. La mayoría de estos pacientes con MF y PV en su historial habían recibido tratamiento previo con hidroxiurea y habían tenido casos previos de CPNM o lesiones precancerosas de la piel. Se recomienda a los pacientes con un mayor riesgo de cáncer de piel que se realicen exámenes de la piel de forma periódica.
GRUPOS ESPECIALES DE PACIENTES
PACIENTES CON DISFUNCIÓN RENAL
La dosis inicial del medicamento JAKAVI debe reducirse para los pacientes con disfunción renal grave. Para los pacientes con MF y enfermedad renal terminal en diálisis, la dosis inicial del medicamento se debe determinar según el recuento de plaquetas, mientras que la dosis inicial recomendada para los pacientes con PV es de 10 mg una vez al día (ver sección "Modo de administración y dosis"). Los pacientes deben estar bajo control estricto en cuanto a la seguridad y la eficacia durante el tratamiento con JAKAVI.
Solo hay datos limitados sobre la dosificación para los pacientes con enfermedad renal terminal en diálisis.
La modelización farmacocinética/farmacodinámica basada en los datos disponibles sugiere que la dosis inicial del medicamento para los pacientes con MF y enfermedad renal terminal en diálisis es de 15-20 mg una vez al día o dos dosis de 10 mg cada 12 horas, administradas después de la diálisis y solo en el día de la diálisis. Para los pacientes con MF y un recuento de plaquetas en el rango de 100.000/μL a 200.000/μL (100 x 10^9/L a 200 x 10^9/L), se prescribe una dosis de 15 mg. Para los pacientes con MF y un recuento de plaquetas superior a 200.000/μL (>200 x 10^9/L), se prescribe una dosis de 20 mg una vez al día o dos dosis de 10 mg cada 12 horas. Las dosis siguientes del medicamento se deben administrar solo en los días de diálisis, después de completar cada sesión de diálisis (una dosis única o dos dosis de 10 mg cada 12 horas).
La dosis inicial recomendada para los pacientes con PV y enfermedad renal terminal en diálisis es de 10 mg una vez al día o 5 mg dos veces al día cada 12 horas, administradas después de la diálisis y solo en el día de la diálisis. Estas recomendaciones de dosificación se basan en la modelización, y cualquier modificación de la dosis para los pacientes con enfermedad renal terminal debe realizarse con un control estricto de la seguridad y la eficacia del medicamento (ver secciones "Modo de administración y dosis" y "Farmacocinética").
No hay datos sobre los pacientes con TTP y enfermedad renal terminal.
PACIENTES CON DISFUNCIÓN HEPÁTICA
Para los pacientes con MF y cualquier grado de disfunción hepática, la dosis inicial del medicamento JAKAVI debe reducirse aproximadamente en un 50% y administrarse dos veces al día. Las dosis siguientes del medicamento se deben ajustar según la seguridad y la eficacia. La dosis inicial recomendada para los pacientes con PV es de 5 mg dos veces al día. Los pacientes con disfunción hepática deben realizarse un análisis de sangre completo, incluyendo un recuento diferencial de leucocitos, al menos cada una o dos semanas durante las primeras seis semanas después del inicio del tratamiento con JAKAVI, y según las indicaciones clínicas, mientras la función hepática y los parámetros sanguíneos se estabilizan. La dosis del medicamento JAKAVI se puede ajustar para minimizar el riesgo de citopenia.
Para los pacientes con disfunción hepática leve, moderada o grave no relacionada con TTP, la dosis inicial de ruxolitinib debe reducirse en un 50% (ver sección "Farmacocinética").
INTERACCIONES
Si JAKAVI se administra concomitantemente con inhibidores potentes de CYP3A4 o inhibidores dobles de CYP2C9 y CYP3A4 (por ejemplo, fluconazol), la dosis estándar de JAKAVI debe reducirse aproximadamente en un 50% y administrarse dos veces al día (ver sección "Interacción con otros medicamentos y otros tipos de interacciones").
Se debe evitar la administración de ruxolitinib con fluconazol en dosis que excedan los 200 mg al día.
La administración concomitante de JAKAVI con terapia citoreductiva se ha asociado con citopenia tratable (ver sección "Modo de administración y dosis").
SÍNDROME DE ABSTINENCIA DEL MEDICAMENTO
Los síntomas de MF pueden regresar dentro de aproximadamente una semana después de la suspensión temporal o la interrupción del tratamiento con JAKAVI. Se han informado casos de abstinencia del medicamento JAKAVI en pacientes que experimentaron reacciones adversas graves, en particular, en presencia de una enfermedad aguda concomitante. Actualmente, no se ha establecido si la interrupción repentina del tratamiento con JAKAVI contribuye al desarrollo de estos fenómenos. Además de los casos en los que la interrupción repentina del tratamiento es necesaria, se debe considerar la posibilidad de reducir gradualmente la dosis de JAKAVI, aunque el beneficio de esta reducción gradual no ha sido demostrado.
EXCIPIENTES
JAKAVI contiene lactosa. Los pacientes con intolerancia conocida a algunos azúcares deben consultar con su médico antes de tomar este medicamento. Los pacientes con enfermedades raras hereditarias, como la intolerancia a la galactosa, la deficiencia grave de lactasa o la mala absorción de glucosa-galactosa, no deben tomar este medicamento.
EMBARAZO Y LACTANCIA
EMBARAZO
No hay datos disponibles sobre la administración del medicamento JAKAVI en mujeres embarazadas. Los estudios en animales han demostrado que el ruxolitinib es embriotóxico y fetotóxico. No se ha observado teratogenicidad en ratas o conejos. Sin embargo, los límites de exposición y la dosis clínica más alta fueron bajos, por lo que la interpretación de los resultados en términos de seguridad para humanos es limitada. El riesgo potencial para humanos es desconocido. Como medida de precaución, la administración del medicamento JAKAVI durante el embarazo está contraindicada (ver sección "Contraindicaciones").
MUJERES EN EDAD REPRODUCTIVA/ANTICONCEPCIÓN
Las mujeres en edad reproductiva deben utilizar métodos anticonceptivos efectivos durante el tratamiento con JAKAVI. En caso de embarazo durante el tratamiento con JAKAVI, se debe evaluar la relación beneficio/riesgo de forma individualizada, considerando cuidadosamente los posibles riesgos para el feto.
LACTANCIA
No se debe administrar JAKAVI durante la lactancia, por lo que se debe suspender la lactancia materna si se inicia el tratamiento. No se sabe si el ruxolitinib y/o sus metabolitos se excretan en la leche materna humana. El riesgo para el lactante no puede ser excluido. Los datos farmacodinámicos/toxicológicos de los estudios en animales demostraron que el ruxolitinib y sus metabolitos se excretan en la leche materna de los animales.
FERTILIDAD
No hay datos disponibles sobre el efecto del ruxolitinib en la fertilidad humana. No se ha observado un efecto en la fertilidad en los estudios en animales.
CAPACIDAD PARA CONDUCIR VEHÍCULOS Y UTILIZAR MAQUINARIA
JAKAVI no tiene o tiene un efecto sedante mínimo. Sin embargo, los pacientes que experimenten mareo después de tomar JAKAVI deben abstenerse de conducir vehículos o operar maquinaria.
MODO DE ADMINISTRACIÓN Y DOSIS
El medicamento JAKAVI se debe administrar por vía oral, con o sin alimentos.
Si se omite una dosis del medicamento, el paciente no debe tomar una dosis adicional, y la siguiente dosis se debe tomar en el momento habitual.
El tratamiento con JAKAVI debe ser iniciado por un médico con experiencia en el uso de medicamentos antineoplásicos.
Antes de iniciar el tratamiento con JAKAVI, se debe realizar un análisis de sangre completo, incluyendo un recuento diferencial de leucocitos.
Los parámetros del análisis de sangre, incluyendo el recuento diferencial de leucocitos, se deben controlar cada 2-4 semanas, y luego según las indicaciones clínicas, hasta la estabilización de la dosis del medicamento JAKAVI (ver sección "Características especiales").
DOSIFICACIÓN
Dosis inicial
La dosis recomendada de JAKAVI para la mielofibrosis (MF) se establece según el recuento de plaquetas (ver tabla "Dosis iniciales para la mielofibrosis").
| Recuento de plaquetas | Dosis inicial |
| Más de 200.000/μL (>200 x 10^9/L) | 20 mg por vía oral dos veces al día |
| De 100.000 a 200.000/μL (100 x 10^9/L a 200 x 10^9/L) | 15 mg por vía oral dos veces al día |
| De 75.000 a menos de 100.000/μL (75 x 10^9/L a <100 x 10^9/L) | 10 mg por vía oral dos veces al día |
| De 50.000 a menos de 75.000/μL (50 x 10^9/L a <75 x 10^9/L) | 5 mg por vía oral dos veces al día |
La dosis recomendada de JAKAVI para los pacientes con policitemia vera (PV) es de 10 mg por vía oral dos veces al día.
La dosis recomendada de JAKAVI para la reacción aguda y crónica de "trasplante contra huésped" (TTPH) es de 10 mg por vía oral dos veces al día. JAKAVI se puede administrar como adyuvante a la terapia de mantenimiento con corticosteroides y/o inhibidores de la calcineurina (ICN).
AJUSTE DE LA DOSIS
Las dosis se pueden ajustar según la seguridad y la eficacia del medicamento.
Mielofibrosis y policitemia vera
Si la eficacia del tratamiento se considera insuficiente, pero los parámetros sanguíneos están en un nivel adecuado, se pueden aumentar las dosis del medicamento de 5 mg dos veces al día a una dosis máxima de 25 mg dos veces al día.
No se debe aumentar la dosis inicial del medicamento durante las primeras cuatro semanas de tratamiento, y luego se puede aumentar no más de cada 2 semanas.
El tratamiento debe suspenderse si el recuento de plaquetas es inferior a 50.000/μL (50 x 10^9/L) o el recuento absoluto de neutrófilos es inferior a 500/μL (0,5 x 10^9/L). En el caso de la PV, el tratamiento también se debe suspender si el nivel de hemoglobina es inferior a 8 g/dL (80 g/L). Después de que los parámetros sanguíneos se estabilicen por encima de estos niveles, se puede reanudar el tratamiento con una dosis de 5 mg dos veces al día, con un aumento gradual de la dosis según el control cuidadoso de los parámetros del análisis de sangre, incluyendo el recuento diferencial de leucocitos.
Se debe reducir la dosis si, durante el tratamiento, el recuento de plaquetas disminuye, como se indica en la tabla "Recomendaciones para la dosificación en pacientes con MF y trombocitopenia", con el fin de evitar la suspensión del tratamiento por trombocitopenia.
| Recuento de plaquetas | Dosis en el momento de la disminución de las plaquetas |
| De 100.000 a menos de 125.000/μL (100 x 10^9/L a 125 x 10^9/L) | 20 mg dos veces al día |
| De 75.000 a menos de 100.000/μL (75 x 10^9/L a 100 x 10^9/L) | 15 mg dos veces al día |
| De 50.000 a menos de 75.000/μL (50 x 10^9/L a 75 x 10^9/L) | 10 mg dos veces al día |
| Menos de 50.000/μL (<50 x 10^9/L) | Suspender el tratamiento |
Reacción de "trasplante contra huésped"
Se debe considerar la reducción de la dosis y la suspensión del tratamiento en pacientes con TTPH y trombocitopenia, neutropenia o aumento del nivel de bilirrubina total después de la terapia de mantenimiento estándar, incluyendo factores de crecimiento, medicamentos antiinfecciosos y transfusiones de sangre. Se recomienda reducir la dosis en un nivel (de 10 mg dos veces al día a 5 mg dos veces al día o de 5 mg dos veces al día a 5 mg una vez al día). A los pacientes que no toleran JAKAVI a una dosis de 5 mg una vez al día, se debe suspender el tratamiento. Las recomendaciones detalladas para la dosificación se presentan en la tabla "Recomendaciones para la dosificación durante la terapia con ruxolitinib en pacientes con TTPH y trombocitopenia, neutropenia o aumento del nivel de bilirrubina total".
| Parámetro de laboratorio | Recomendaciones para la dosificación |
| Recuento de plaquetas < 20.000/μL (<20 x 10^9/L) | Reducir la dosis de JAKAVI en un nivel. Si el recuento de plaquetas permanece ≥ 20.000/μL (≥20 x 10^9/L) durante 7 días, se puede aumentar la dosis al nivel inicial; de lo contrario, se debe mantener la dosis reducida. |
| Recuento de plaquetas < 15.000/μL (<15 x 10^9/L) | Suspender la administración del medicamento JAKAVI hasta que el recuento de plaquetas sea ≥ 20.000/μL (≥20 x 10^9/L), y luego reanudar la administración con una dosis reducida en un nivel. |
| Recuento absoluto de neutrófilos (RAN) de ≥ 500/μL a < 750/μL (0,5 x 10^9/L a 0,75 x 10^9/L) | Reducir la dosis de JAKAVI en un nivel. Luego, se puede reanudar la administración con la dosis inicial si el RAN es > 1.000/μL (>1 x 10^9/L). |
| Recuento absoluto de neutrófilos < 500/μL (<0,5 x 10^9/L) | Suspender la administración de JAKAVI hasta que el RAN sea > 500/μL (>0,5 x 10^9/L), y luego reanudar la administración con una dosis reducida en un nivel. Si el RAN es > 1.000/μL (>1 x 10^9/L), se puede reanudar la administración con la dosis inicial. |
AJUSTE DE LA DOSIS DEL MEDICAMENTO EN CASO DE ADMINISTRACIÓN CONCOMITANTE CON INHIBIDORES POTENTES DE CYP3A4 O INHIBIDORES DOBLES DE CYP2C9/3A4
Cuando se administra ruxolitinib con inhibidores potentes de CYP3A4 o inhibidores dobles de CYP2C9 y CYP3A4 (por ejemplo, fluconazol), se debe reducir la dosis estándar de ruxolitinib aproximadamente en un 50% y administrarla dos veces al día (ver sección "Interacción con otros medicamentos y otros tipos de interacciones").
Se debe evitar la administración de ruxolitinib con fluconazol en dosis que excedan los 200 mg al día.
Se recomienda un control más frecuente (por ejemplo, dos veces a la semana) de los parámetros hematológicos y los síntomas clínicos de las reacciones adversas relacionadas con JAKAVI durante la administración concomitante con inhibidores potentes de CYP3A4 o inhibidores dobles de CYP2C9 y CYP3A4.
GRUPOS ESPECIALES DE PACIENTES
Disfunción renal
No se requiere ajuste de la dosis del medicamento para los pacientes con disfunción renal leve o moderada.
Para los pacientes con disfunción renal grave (aclaramiento de creatinina < 30 mL/min), se debe reducir la dosis inicial del medicamento según el recuento de plaquetas en MF aproximadamente en un 50% y administrarla dos veces al día. La dosis inicial recomendada para los pacientes con PV es de 5 mg dos veces al día. Con respecto a la seguridad y la eficacia, los pacientes deben estar bajo control estricto durante el tratamiento con JAKAVI.
Solo hay datos limitados sobre la dosificación para los pacientes con enfermedad renal terminal en diálisis.
La modelización farmacocinética/farmacodinámica basada en los datos disponibles sugiere que la dosis inicial del medicamento para los pacientes con MF y enfermedad renal terminal en diálisis es de 15-20 mg una vez al día o dos dosis de 10 mg cada 12 horas, administradas después de la diálisis y solo en el día de la diálisis. La dosis inicial recomendada para los pacientes con PV es de 10 mg una vez al día o 5 mg dos veces al día cada 12 horas, administradas después de la diálisis y solo en el día de la diálisis. Estas recomendaciones de dosificación se basan en la modelización, y cualquier modificación de la dosis para los pacientes con enfermedad renal terminal debe realizarse con un control estricto de la seguridad y la eficacia del medicamento (ver secciones "Modo de administración y dosis" y "Farmacocinética").
No hay datos sobre los pacientes con TTP y enfermedad renal terminal.
Disfunción hepática
Para los pacientes con MF y cualquier grado de disfunción hepática, la dosis inicial del medicamento JAKAVI debe reducirse aproximadamente en un 50% y administrarse dos veces al día. Las dosis siguientes del medicamento se deben ajustar según la seguridad y la eficacia. La dosis inicial recomendada para los pacientes con PV es de 5 mg dos veces al día. Los pacientes con disfunción hepática deben realizarse un análisis de sangre completo, incluyendo un recuento diferencial de leucocitos, al menos cada una o dos semanas durante las primeras seis semanas después del inicio del tratamiento con JAKAVI, y según las indicaciones clínicas, mientras la función hepática y los parámetros sanguíneos se estabilizan. La dosis del medicamento JAKAVI se puede ajustar para minimizar el riesgo de citopenia.
Para los pacientes con disfunción hepática leve, moderada o grave no relacionada con TTP, la dosis inicial de ruxolitinib debe reducirse en un 50% (ver sección "Farmacocinética").
PACIENTES DE EDAD AVANZADA (≥ 65 AÑOS)
No se requiere ajuste adicional de la dosis del medicamento para los pacientes de edad avanzada.
SUSPENSIÓN DEL TRATAMIENTO
El tratamiento de MF y PV con JAKAVI se puede prolongar mientras la relación beneficio/riesgo sea favorable. Sin embargo, el tratamiento se debe suspender después de 6 meses si no se observa una reducción del tamaño del bazo o un alivio de los síntomas después del inicio del tratamiento.
Se recomienda suspender el tratamiento con ruxolitinib en pacientes que han demostrado algún grado de mejora clínica si experimentan un aumento adicional del tamaño del bazo del 40% en comparación con el tamaño inicial (lo que equivale aproximadamente a un aumento del 25% del volumen del bazo) y no experimentan un alivio adicional de los síntomas relacionados con la enfermedad.
En el caso de TTPH, se debe considerar la posibilidad de reducir gradualmente la dosis del medicamento JAKAVI en pacientes que responden al tratamiento y después de suspender la administración de corticosteroides. Se recomienda reducir la dosis del medicamento JAKAVI en un 50% cada 2 meses. Se debe considerar la posibilidad de aumentar la dosis nuevamente si los signos o síntomas de TTPH reaparecen durante o después de la reducción de la dosis de JAKAVI.
NIÑOS
La seguridad y la eficacia del medicamento JAKAVI en niños (menores de 18 años) con MF y PV no se han estudiado. No hay datos disponibles (ver sección "Farmacodinamia").
La seguridad y la eficacia del medicamento JAKAVI en niños de 12 años de edad o más con TTPH se confirman con los datos de los estudios aleatorizados de fase 3 REACH2 y REACH3. La dosis del medicamento JAKAVI para los niños con TTPH es la misma que para los adultos. La seguridad y la eficacia del medicamento JAKAVI en niños menores de 12 años no se han establecido.
SOBREDOSIS
No se conoce un antídoto para la sobredosis del medicamento JAKAVI. La administración de dosis únicas del medicamento de hasta 200 mg se asoció con una tolerabilidad aceptable del medicamento. La administración de dosis más altas del medicamento que las dosis repetidas recomendadas se asoció con un aumento de la mielosupresión, incluyendo leucopenia, anemia y trombocitopenia. En estos casos, se debe aplicar una terapia de apoyo adecuada. No se espera que la diálisis acelere la eliminación del ruxolitinib del organismo.
Efectos adversos
Resumen del perfil de seguridad
Mielofibrosis
Las reacciones adversas más frecuentes informadas fueron trombocitopenia y anemia.
Las reacciones adversas hematológicas (cualquier grado según los Criterios comunes de terminología para eventos adversos [CTCAE]) incluyeron casos de anemia (83,8%), trombocitopenia (80,5%) y neutropenia (20,8%).
La anemia, la trombocitopenia y la neutropenia son efectos dependientes de la dosis.
Las tres reacciones adversas no hematológicas más frecuentes fueron moretones (33,3%), otras hemorragias (incluyendo epistaxis, hemorragias postprocedimiento y hematuria) (24,3%) y mareo (21,9%).
Las tres anomalías de laboratorio no hematológicas más frecuentes fueron niveles elevados de alanina aminotransferasa (40,7%), niveles elevados de aspartato aminotransferasa (31,5%) e hipertrigliceridemia (25,2%). En los estudios de fase 3 en pacientes con MF, no se observaron hipertrigliceridemia ni niveles elevados de aspartato aminotransferasa de grado 3 o 4 según la CTCAE, ni niveles elevados de alanina aminotransferasa o hipercolesterolemia de grado 4 según la CTCAE.
La interrupción del tratamiento debido a reacciones adversas, independientemente de la relación causal, se produjo en el 30,0% de los pacientes.
Policitemia vera
Las reacciones adversas más frecuentes informadas fueron anemia y niveles elevados de alanina aminotransferasa.
Las reacciones adversas hematológicas (cualquier grado según la CTCAE) incluyeron anemia (61,8%), trombocitopenia (25,0%) y neutropenia (5,3%). La anemia y la trombocitopenia de grado 3 o 4 según la CTCAE se produjeron en el 2,9% o el 2,6% de los pacientes, respectivamente.
Las tres reacciones adversas no hematológicas más frecuentes fueron aumento de peso (20,3%), mareo (19,4%) y cefalea (17,9%).
Las tres anomalías de laboratorio no hematológicas más frecuentes identificadas como reacciones adversas fueron niveles elevados de alanina aminotransferasa (45,3%), niveles elevados de aspartato aminotransferasa (42,6%) e hipercolesterolemia (34,7%). No se produjeron niveles elevados de alanina aminotransferasa o hipercolesterolemia de grado 4 según la CTCAE, y se registró un caso de niveles elevados de aspartato aminotransferasa de grado 4 según la CTCAE.
La interrupción del tratamiento debido a reacciones adversas, independientemente de la relación causal, se produjo en el 19,4% de los pacientes.
Trombosis trombocítica trombocitopénica (TTP)
Las reacciones adversas más frecuentes informadas fueron trombocitopenia, anemia y neutropenia.
Las anomalías de laboratorio hematológicas definidas como reacciones adversas incluyeron trombocitopenia (85,2%), anemia (75,0%) y neutropenia (65,1%). La anemia de grado 3 se produjo en el 47,7% de los pacientes (no se prevé un grado 4 según la CTCAE versión 4.03). La trombocitopenia de grado 3 y 4 se produjo en el 31,3% y el 47,7% de los pacientes, respectivamente.
Las tres reacciones adversas no hematológicas más frecuentes incluyeron infección por citomegalovirus (32,3%), sepsis (25,4%) e infecciones del tracto urinario (17,9%).
Las tres anomalías de laboratorio no hematológicas más frecuentes definidas como reacciones adversas incluyeron niveles elevados de alanina aminotransferasa (54,9%), niveles elevados de aspartato aminotransferasa (52,3%) e hipercolesterolemia (49,2%). La mayoría de ellas fueron de grado 1 y 2.
La interrupción del tratamiento debido a eventos adversos, independientemente de la relación causal, se produjo en el 29,4% de los pacientes.
Trombosis trombocítica trombocitopénica crónica (TTPc)
Las reacciones adversas más frecuentes informadas fueron anemia, hipercolesterolemia y niveles elevados de aspartato aminotransferasa.
Las anomalías de laboratorio hematológicas definidas como reacciones adversas incluyeron anemia (68,6%), trombocitopenia (34,4%) y neutropenia (36,2%). La anemia de grado 3 se produjo en el 14,8% de los pacientes (no se prevé un grado 4 según la CTCAE versión 4.03). La neutropenia de grado 3 y 4 se produjo en el 9,5% y el 6,7% de los pacientes, respectivamente.
Las tres reacciones adversas no hematológicas más frecuentes incluyeron hipertensión arterial (15,0%), cefalea (10,2%) e infecciones del tracto urinario (9,3%).
Las tres anomalías de laboratorio no hematológicas más frecuentes definidas como reacciones adversas incluyeron hipercolesterolemia (52,3%), niveles elevados de aspartato aminotransferasa (52,2%) y niveles elevados de alanina aminotransferasa (43,1%). La mayoría de ellas fueron de grado 1 y 2.
La interrupción del tratamiento debido a eventos adversos, independientemente de la relación causal, se produjo en el 18,1% de los pacientes.
Resumen de reacciones adversas durante los estudios clínicos
La seguridad en pacientes con MF se evaluó en función de los datos de seguimiento a largo plazo de dos estudios de fase 3 (COMFORT I y COMFORT II), incluyendo datos de pacientes que inicialmente fueron aleatorizados al grupo de ruxolitinib (n = 301) y de pacientes que recibieron ruxolitinib después de cambiar del grupo de control (n = 156). La mediana de exposición en la que se basan las categorías de frecuencia de reacciones adversas al medicamento (RAM) en pacientes con MF es de 30,5 meses (rango de 0,3 a 68,1 meses).
La seguridad en pacientes con PV se evaluó en función de los datos de seguimiento a largo plazo de dos estudios de fase 3 (RESPONSE, RESPONSE 2), incluyendo datos de pacientes que inicialmente fueron aleatorizados al grupo de ruxolitinib (n = 184) y de pacientes que recibieron ruxolitinib después de cambiar del grupo de control (n = 156). La mediana de exposición en la que se basan las categorías de frecuencia de RAM en pacientes con PV es de 41,7 meses (rango de 0,03 a 59,7 meses).
La seguridad del medicamento Jakavi en pacientes con TTP aguda se evaluó en el estudio de fase 3 REACH2, que incluyó datos de pacientes que inicialmente fueron aleatorizados al grupo de Jakavi (n = 152) y de pacientes que recibieron Jakavi después de cambiar del grupo de mejor tratamiento disponible (MTD) (n = 49). La mediana de exposición en la que se basan las categorías de frecuencia de RAM es de 8,9 semanas (rango de 0,3 a 66,1 semanas).
La seguridad del medicamento Jakavi en pacientes con TTP crónica se evaluó en el estudio de fase 3 REACH3, que incluyó datos de pacientes que inicialmente fueron aleatorizados al grupo de Jakavi (n = 165) y de pacientes que recibieron Jakavi después de cambiar del grupo de mejor tratamiento disponible (MTD) (n = 61). La mediana de exposición en la que se basan las categorías de frecuencia de RAM es de 41,4 semanas (rango de 0,7 a 127,3 semanas).
En el programa de estudios clínicos, la gravedad de las reacciones adversas se evaluó según la CTCAE, que define el grado 1 como leve, el grado 2 como moderado, el grado 3 como grave, el grado 4 como potencialmente mortal o incapacitante, y el grado 5 como mortal.
Frecuencia de reacciones adversas en los estudios de fase 3 en pacientes con MF y PV
| Reacciones adversas | Frecuencia en pacientes con MF | Frecuencia en pacientes con PV |
| Infecciones e invasiones | ||
| Infecciones del tracto urinario | Muy frecuente | Muy frecuente |
| Herpes zóster | Muy frecuente | Muy frecuente |
| Neumonía | Muy frecuente | Frecuente |
| Sepsis | Frecuente | No frecuente |
| Tuberculosis | No frecuente | Desconocido |
| Reactivación del virus de la hepatitis B | Desconocido | No frecuente |
| Trastornos de la sangre y del sistema linfático | ||
| Anemia | ||
| Grado 4 según la CTCAE | Muy frecuente | No frecuente |
| Grado 3 según la CTCAE | Muy frecuente | Frecuente |
| Cualquier grado según la CTCAE | Muy frecuente | Muy frecuente |
| Trombocitopenia | ||
| Grado 4 según la CTCAE | Frecuente | No frecuente |
| Grado 3 según la CTCAE | Muy frecuente | Frecuente |
| Cualquier grado según la CTCAE | Muy frecuente | Muy frecuente |
| Neutropenia | ||
| Grado 4 según la CTCAE | Frecuente | No frecuente |
| Grado 3 según la CTCAE | Frecuente | No frecuente |
| Cualquier grado según la CTCAE | Muy frecuente | Frecuente |
| Pancitopenia | Frecuente | Frecuente |
| Hemorragias (cualquier tipo de hemorragia, incluyendo hemorragias intracraneales, hemorragias gastrointestinales, moretones y otras hemorragias) | Muy frecuente | Muy frecuente |
| Moretones | Muy frecuente | Muy frecuente |
| Hemorragias gastrointestinales | Muy frecuente | Frecuente |
| Hemorragias intracraneales | Frecuente | No frecuente |
| Otras hemorragias (incluyendo epistaxis, hemorragias postprocedimiento y hematuria) | Muy frecuente | Muy frecuente |
| Trastornos del metabolismo y la nutrición | ||
| Hipercolesterolemia | Muy frecuente | Muy frecuente |
| Hipertrigliceridemia | Muy frecuente | Muy frecuente |
| Aumento de peso | Muy frecuente | Muy frecuente |
| Trastornos del sistema nervioso | ||
| Mareo | Muy frecuente | Muy frecuente |
| Cefalea | Muy frecuente | Muy frecuente |
| Trastornos gastrointestinales | ||
| Aumento de la lipasa | Muy frecuente | Muy frecuente |
| Estreñimiento | Muy frecuente | Muy frecuente |
| Flatulencia | Frecuente | Frecuente |
| Trastornos hepáticos y biliares | ||
| Aumento de la alanina aminotransferasa | ||
| Grado 3 según la CTCAE | Frecuente | Frecuente |
| Cualquier grado según la CTCAE | Muy frecuente | Muy frecuente |
| Aumento de la aspartato aminotransferasa | ||
| Cualquier grado según la CTCAE | Muy frecuente | Muy frecuente |
| Trastornos cardiovasculares | ||
| Hipertensión arterial | Muy frecuente | Muy frecuente |
Frecuencia de reacciones adversas en los estudios de fase 3 en pacientes con TTP
| Reacciones adversas | TTP aguda (REACH2) | TTP crónica (REACH3) |
| Infecciones e invasiones | ||
| Infecciones por citomegalovirus | Muy frecuente | Frecuente |
| Grado ≥ 3 según la CTCAE | Muy frecuente | Frecuente |
| Sepsis | Muy frecuente | - |
| Grado ≥ 3 según la CTCAE | Muy frecuente | - |
| Infecciones del tracto urinario | Muy frecuente | Frecuente |
| Grado ≥ 3 según la CTCAE | Frecuente | Frecuente |
| Infecciones virales BK | - | Frecuente |
| Grado ≥ 3 según la CTCAE | - | No frecuente |
| Trastornos de la sangre y del sistema linfático | ||
| Trombocitopenia | Muy frecuente | Muy frecuente |
| Grado 3 según la CTCAE | Muy frecuente | Frecuente |
| Grado 4 según la CTCAE | Muy frecuente | Muy frecuente |
| Anemia | Muy frecuente | Muy frecuente |
| Grado 3 según la CTCAE | Muy frecuente | Muy frecuente |
| Neutropenia | Muy frecuente | Muy frecuente |
| Grado 3 según la CTCAE | Muy frecuente | Frecuente |
| Grado 4 según la CTCAE | Muy frecuente | Frecuente |
| Pancitopenia | Muy frecuente | - |
| Trastornos del metabolismo y la nutrición | ||
| Hipercolesterolemia | Muy frecuente | Muy frecuente |
| Grado 3 según la CTCAE | Frecuente | Frecuente |
| Grado 4 según la CTCAE | Frecuente | No frecuente |
| Aumento de peso | - | Frecuente |
| Grado ≥ 3 según la CTCAE | - | Datos no disponibles |
| Trastornos del sistema nervioso | ||
| Cefalea | Frecuente | Muy frecuente |
| Grado ≥ 3 según la CTCAE | No frecuente | Frecuente |
| Trastornos cardiovasculares | ||
| Hipertensión arterial | Muy frecuente | Muy frecuente |
| Grado ≥ 3 según la CTCAE | Frecuente | Frecuente |
| Trastornos gastrointestinales | ||
| Aumento de la lipasa | - | Muy frecuente |
| Grado 3 según la CTCAE | - | Frecuente |
| Grado 4 según la CTCAE | - | No frecuente |
| Aumento de la amilasa | - | Muy frecuente |
| Grado 3 según la CTCAE | - | Frecuente |
| Grado 4 según la CTCAE | - | Frecuente |
| Náuseas | Muy frecuente | - |
| Grado ≥ 3 según la CTCAE | No frecuente | - |
| Estreñimiento | - | Frecuente |
| Grado ≥ 3 según la CTCAE | - | Datos no disponibles |
| Trastornos hepáticos y biliares | ||
| Aumento de la alanina aminotransferasa | Muy frecuente | Muy frecuente |
| Grado 3 según la CTCAE | Muy frecuente | Frecuente |
| Grado 4 según la CTCAE | Frecuente | No frecuente |
| Aumento de la aspartato aminotransferasa | Muy frecuente | Muy frecuente |
| Grado 3 según la CTCAE | Frecuente | Frecuente |
| Grado 4 según la CTCAE | Datos no disponibles | No frecuente |
| Trastornos musculoesqueléticos y del tejido conjuntivo | ||
| Aumento de la creatina quinasa en sangre | - | Muy frecuente |
| Grado 3 según la CTCAE | - | Frecuente |
| Grado 4 según la CTCAE | - | Frecuente |
| Trastornos renales y urinarios | ||
| Aumento de la creatinina en sangre | - | Muy frecuente |
| Grado 3 según la CTCAE | - | Frecuente |
| Grado 4 según la CTCAE | - | Datos no disponibles |
| 1 La frecuencia se define según los nuevos o empeorados desviaciones de los parámetros de laboratorio desde los valores normales en comparación con los niveles basales. | ||
Descripción de reacciones adversas individuales
Anemia
En los estudios clínicos de fase 3 en pacientes con MF, la mediana del tiempo hasta la primera aparición de anemia de grado 2 o superior según la CTCAE fue de 1,5 meses. Un paciente (0,3%) interrumpió el tratamiento debido al desarrollo de anemia.
En pacientes que recibieron ruxolitinib, el valor medio de la disminución máxima del nivel de hemoglobina desde el nivel basal fue de 10 g/L después de 8-12 semanas de tratamiento, tras lo cual el nivel de hemoglobina se recuperó gradualmente hasta un nuevo nivel de equilibrio, que fue aproximadamente 5 g/L menor que el nivel basal. Esta tendencia se observó en pacientes independientemente de si recibieron o no transfusiones durante el tratamiento.
En el estudio aleatorizado y controlado con placebo COMFORT I, el 60,6% de los pacientes con MF que recibieron ruxolitinib y el 37,7% de los pacientes con MF que recibieron placebo recibieron transfusiones de masas eritrocitarias durante el período de tratamiento aleatorizado. En el estudio COMFORT II, la frecuencia de transfusiones de masas eritrocitarias fue del 53,4% en el grupo de ruxolitinib y del 41,1% en el grupo de tratamiento óptimo disponible.
En el período aleatorizado de los estudios de fase 3, se informó menos frecuentemente de anemia en pacientes con PV (40,8%) en comparación con pacientes con MF (82,4%). En la población de pacientes con PV, se informó de eventos de grado 3 y 4 según la CTCAE en el 2,7%, mientras que en la población de pacientes con MF, la frecuencia fue del 42,56%.
Durante los estudios de fase 3 en pacientes con TTP aguda y crónica, se informó de anemia de grado 3 según la CTCAE en el 47,7% y el 14,8% de los pacientes, respectivamente.
Trombocitopenia
En los estudios clínicos de fase 3 en pacientes con MF, en los que se desarrolló trombocitopenia de grado 3 o 4, la mediana del tiempo hasta la primera aparición fue de aproximadamente 8 semanas. En general, la trombocitopenia fue reversible después de la disminución de la dosis o la interrupción del tratamiento. La mediana del tiempo hasta la recuperación de la cuenta de plaquetas por encima de 50.000/μL fue de 14 días. En el período aleatorizado, se transfundieron masas plaquetarias al 4,7% de los pacientes que recibieron ruxolitinib y al 4,0% de los pacientes que recibieron esquemas de tratamiento de control. La interrupción del tratamiento debido a trombocitopenia se produjo en el 0,7% de los pacientes que recibieron ruxolitinib y en el 0,9% de los pacientes que recibieron esquemas de tratamiento de control. En pacientes con una cuenta de plaquetas antes del tratamiento entre 100.000 y 200.000/μL, hubo una frecuencia más alta de trombocitopenia de grado 3 o 4 en comparación con pacientes con una cuenta de plaquetas > 200.000/μL (64,2% en comparación con 38,5%).
En el período aleatorizado de los estudios de fase 3, la trombocitopenia se produjo con menos frecuencia en pacientes con PV (16,8%) en comparación con pacientes con MF (69,8%). La frecuencia de casos graves (es decir, de grado 3 o 4 según la CTCAE) de trombocitopenia en pacientes con PV (2,7%) fue menor que en pacientes con MF (11,6%).
En el estudio de fase 3 en pacientes con TTP aguda, la trombocitopenia de grado 3 y 4 se produjo en el 31,3% y el 47,7% de los pacientes, respectivamente. En el estudio de fase 3 en pacientes con TTP crónica, la frecuencia de desarrollo de trombocitopenia de grado 3 y 4 fue menor (5,9% y 10,7%, respectivamente) que en pacientes con TTP aguda.
Neutropenia
En los estudios clínicos de fase 3 en pacientes con MF, en los que se desarrolló neutropenia de grado 3 o 4, la mediana del tiempo hasta la primera aparición fue de aproximadamente 12 semanas. En el período aleatorizado, se informó de la interrupción del tratamiento o la disminución de la dosis debido a neutropenia en el 1,0% de los pacientes, y se interrumpió el tratamiento debido a neutropenia en el 0,3% de los pacientes.
En el período aleatorizado de los estudios de fase 3 en pacientes con PV, se informó de neutropenia en el 1,6% de los pacientes que recibieron ruxolitinib en comparación con el 7% de los pacientes del grupo de control. En el grupo de ruxolitinib, un paciente desarrolló neutropenia de grado 4 según la CTCAE. Durante el seguimiento a largo plazo, se informó de neutropenia de grado 4 según la CTCAE en 2 pacientes.
En los estudios clínicos de fase 3 en pacientes con TTP aguda, la neutropenia de grado 3 o 4 se produjo en el 17,9% y el 20,6% de los pacientes, respectivamente. En los estudios clínicos de fase 3 en pacientes con TTP crónica, la frecuencia de desarrollo de neutropenia de grado 3 o 4 fue menor (9,5% y 6,7%, respectivamente) que en pacientes con TTP aguda.
Hemorragias
En los estudios de fase 3 en pacientes con MF, se informó de casos de hemorragia (incluyendo hemorragias intracraneales y hemorragias gastrointestinales, moretones y otras hemorragias) en el 32,6% de los pacientes que recibieron ruxolitinib y en el 23,2% de los pacientes que recibieron tratamiento de control (placebo o tratamiento óptimo disponible). La frecuencia de eventos de grado 3-4 fue similar en pacientes que recibieron ruxolitinib o tratamiento de control (4,7% en comparación con 3,1%). La mayoría de los pacientes con hemorragia durante el tratamiento informaron de moretones (65,3%). Los moretones se informaron con más frecuencia en pacientes que recibieron ruxolitinib en comparación con aquellos que recibieron tratamiento de control (21,3% en comparación con 11,6%). Se informó de hemorragias intracraneales en el 1% de los pacientes que recibieron ruxolitinib y en el 0,9% de los pacientes que recibieron tratamiento de control. Se informó de hemorragias gastrointestinales en el 5,0% de los pacientes que recibieron ruxolitinib en comparación con el 3,1% en el tratamiento de control. Se informó de otras hemorragias (incluyendo epistaxis, hemorragias postprocedimiento y hematuria) en el 13,3% de los pacientes que recibieron ruxolitinib y en el 10,3% en el tratamiento de control.
Durante el seguimiento a largo plazo en los estudios clínicos de fase 3 en pacientes con MF, la frecuencia total de casos de hemorragia aumentó proporcionalmente con el aumento del tiempo de seguimiento. La formación de moretones es el caso de hemorragia más frecuentemente informado (33,3%). Las hemorragias intracraneales y las hemorragias gastrointestinales se produjeron en el 1,3% y el 10,1% de los pacientes, respectivamente.
En el período aleatorizado de los estudios de fase 3 en pacientes con PV, se informó de casos de hemorragia (incluyendo hemorragias intracraneales y hemorragias gastrointestinales, moretones y otras hemorragias) en el 16,8% de los pacientes que recibieron ruxolitinib, en el 15,3% de los pacientes que recibieron tratamiento óptimo disponible en el estudio RESPONSE y en el 12,0% de los pacientes que recibieron tratamiento óptimo disponible en el estudio RESPONSE 2. Se informó de moretones en el 10,3% de los pacientes que recibieron ruxolitinib, en el 8,1% de los pacientes que recibieron tratamiento óptimo disponible en el estudio RESPONSE y en el 2,7% de los pacientes que recibieron tratamiento óptimo disponible en el estudio RESPONSE 2. No se informó de hemorragias intracraneales ni de hemorragias gastrointestinales en pacientes que recibieron ruxolitinib. Un paciente que recibió ruxolitinib tuvo un caso de hemorragia de grado 3 (hemorragia postprocedimiento); no se informó de casos de hemorragia de grado 4. Se informó de otras hemorragias (incluyendo epistaxis, hemorragias postprocedimiento y hemorragias nasales) en el 8,7% de los pacientes que recibieron ruxolitinib, en el 6,3% de los pacientes que recibieron tratamiento óptimo disponible en el estudio RESPONSE y en el 6,7% de los pacientes que recibieron tratamiento óptimo disponible en el estudio RESPONSE 2.
Durante el seguimiento a largo plazo en los estudios de fase 3 en pacientes con PV, la frecuencia total de casos de hemorragia aumentó proporcionalmente con el aumento del tiempo de seguimiento. La formación de moretones es el caso de hemorragia más frecuentemente informado (17,4%). Las hemorragias intracraneales y las hemorragias gastrointestinales se produjeron en el 0,3% y el 3,5% de los pacientes, respectivamente.
En el período aleatorizado del estudio de fase 3 en pacientes con TTP aguda, los casos de hemorragia se produjeron en el 25,0% y el 22,0% de los pacientes en los grupos de ruxolitinib y MTD, respectivamente. Los casos de hemorragia fueron generalmente similares en pacientes de ambos grupos de tratamiento: moretones (5,9% en el grupo de ruxolitinib en comparación con 6,7% en el grupo de MTD), hemorragias gastrointestinales (9,2% en comparación con 6,7%) y otras hemorragias (13,2% en comparación con 10,7%). Se produjeron hemorragias intracraneales en el 0,7% de los pacientes en el grupo de MTD y no se produjeron en ningún paciente en el grupo de ruxolitinib.
En el período aleatorizado del estudio de fase 3 en pacientes con TTP crónica, los casos de hemorragia se produjeron en el 11,5% y el 14,6% de los pacientes en los grupos de ruxolitinib y MTD, respectivamente. Los casos de hemorragia fueron generalmente similares en pacientes de ambos grupos de tratamiento: moretones (4,2% en el grupo de ruxolitinib en comparación con 2,5% en el grupo de MTD), hemorragias gastrointestinales (1,2% en comparación con 3,2%) y otras hemorragias (6,7% en comparación con 10,1%). No se produjeron hemorragias intracraneales en ningún grupo de tratamiento.
Infecciones
En los estudios de fase 3 en pacientes con MF, se informó de infección del tracto urinario de grado 3 o 4 en el 1,0% de los pacientes, de herpes zóster en el 4,3% y de tuberculosis en el 1,0%. En los estudios clínicos de fase 3, se informó de sepsis en el 3,0% de los pacientes. Los resultados del seguimiento extendido en pacientes que recibieron ruxolitinib no mostraron tendencia a un aumento en la frecuencia de sepsis con el tiempo.
En el período aleatorizado de los estudios de fase 3 en pacientes con PV, se informó de infección del tracto urinario de grado 3 según la CTCAE en un caso (0,5%), y no hubo casos de grado 4. La frecuencia de herpes zóster fue similar en pacientes con PV (4,3%) y pacientes con MF (4,0%). Hubo un informe de neuralgia postherpética de grado 3 según la CTCAE en pacientes con PV. La neumonía se produjo en el 0,5% de los pacientes que recibieron ruxolitinib en comparación con el 1,6% de los pacientes que recibieron tratamiento de control. Ningún paciente en el grupo de ruxolitinib informó de sepsis o tuberculosis.
Durante el seguimiento a largo plazo en los estudios de fase 3 en pacientes con PV, se informó frecuentemente de infecciones del tracto urinario (11,8%), herpes zóster (14,7%) y neumonía (7,1%). La sepsis se registró en el 0,6% de los pacientes. Durante el seguimiento, no se produjo tuberculosis en ningún paciente.
En el período aleatorizado del estudio de fase 3 en pacientes con TTP aguda, las infecciones del tracto urinario se produjeron en el 9,9% (grado ≥ 3, 3,3%) de los pacientes en el grupo de ruxolitinib en comparación con el 10,7% (grado ≥ 3, 6,0%) en el grupo de MTD. Las infecciones por citomegalovirus se produjeron en el 28,3% (grado ≥ 3, 9,3%) de los pacientes en el grupo de ruxolitinib en comparación con el 24,0% (grado ≥ 3, 10,0%) en el grupo de MTD. Los casos de sepsis se produjeron en el 12,5% (grado ≥ 3, 11,1%) de los pacientes en el grupo de ruxolitinib en comparación con el 8,7% (grado ≥ 3, 6,0%) en el grupo de MTD. La infección viral BK se produjo solo en el grupo de ruxolitinib en 3 pacientes, con un caso de grado 3. Durante el seguimiento extendido en pacientes que recibieron ruxolitinib, las infecciones del tracto urinario se produjeron en el 17,9% (grado ≥ 3, 6,5%) de los pacientes, y las infecciones por citomegalovirus se produjeron en el 32,3% (grado ≥ 3, 11,4%) de los pacientes. Las infecciones por citomegalovirus con afectación de órganos se produjeron en muy pocos pacientes; la colitis por citomegalovirus, la enteritis por citomegalovirus y la infección por citomegalovirus del tracto gastrointestinal de cualquier grado se produjeron en 4, 2 y 1 paciente, respectivamente. Los casos de sepsis, incluyendo el choque séptico, de cualquier grado se produjeron en el 25,4% (grado ≥ 3, 21,9%) de los pacientes.
En el período aleatorizado del estudio de fase 3 en pacientes con TTP crónica, las infecciones del tracto urinario se produjeron en el 8,5% (grado ≥ 3, 1,2%) de los pacientes en el grupo de ruxolitinib en comparación con el 6,3% (grado ≥ 3, 1,3%) en el grupo de MTD. La infección viral BK (Betapolyomavirus) se produjo en el 5,5% (grado ≥ 3, 0,6%) de los pacientes en el grupo de ruxolitinib en comparación con el 1,3% en el grupo de MTD. Las infecciones por citomegalovirus se produjeron en el 9,1% (grado ≥ 3, 1,8%) de los pacientes en el grupo de ruxolitinib en comparación con el 10,8% (grado ≥ 3, 1,9%) en el grupo de MTD. Los casos de sepsis se produjeron en el 2,4% (grado ≥ 3, 2,4%) de los pacientes en el grupo de ruxolitinib en comparación con el 6,3% (grado ≥ 3, 5,7%) en el grupo de MTD. Durante el seguimiento extendido en pacientes que recibieron ruxolitinib, las infecciones del tracto urinario y las infecciones virales BK se produjeron en el 9,3% (grado ≥ 3, 1,3%) y el 4,9% (grado ≥ 3, 0,4%) de los pacientes, respectivamente. Las infecciones por citomegalovirus y los casos de sepsis se produjeron en el 8,8% (grado ≥ 3, 1,3%) y el 3,5% (grado ≥ 3, 3,5%) de los pacientes, respectivamente.
Aumento de la lipasa
En el período aleatorizado del estudio RESPONSE, la disminución de los niveles de lipasa fue mayor en el grupo de ruxolitinib en comparación con el grupo de control, principalmente debido a la diferencia en los aumentos de grado 1 (18,2% en comparación con 8,1%). Los aumentos de grado ≥ 2 fueron similares en los grupos de tratamiento. En el estudio RESPONSE 2, la frecuencia fue comparable en el grupo de ruxolitinib y el grupo de control (10,8% en comparación con 8%). Durante el seguimiento a largo plazo en los estudios de fase 3 en pacientes con PV, se informó de aumento de la lipasa de grado 3 y 4 en el 7,4% y el 0,9% de los pacientes, respectivamente. En estos pacientes, no se informó de signos y síntomas de pancreatitis ni de aumento de la lipasa.
En los estudios de fase 3 en pacientes con MF, los niveles altos de lipasa se produjeron en el 18,7% y el 19,3% de los pacientes en el grupo de ruxolitinib en comparación con el 16,6% y el 14,0% en los grupos de control durante los estudios COMFORT I y COMFORT II. No se informó de signos y síntomas de pancreatitis en pacientes con aumento de la lipasa.
En el período aleatorizado del estudio de fase 3 en pacientes con TTP aguda, los nuevos o empeorados valores de lipasa se registraron en el 19,7% de los pacientes en el grupo de ruxolitinib en comparación con el 12,5% en el grupo de MTD; el aumento correspondiente en casos de grado 3 y 4 fue similar (3,1% en comparación con 5,1%). Durante el seguimiento extendido en pacientes que recibieron ruxolitinib, los valores de lipasa aumentada se produjeron en el 32,2% de los pacientes; la enfermedad de grado 3 y 4 se produjo en el 8,7% y el 2,2% de los pacientes, respectivamente.
En el período aleatorizado del estudio de fase 3 en pacientes con TTP crónica, los nuevos o empeorados valores de lipasa se produjeron en el 32,1% de los pacientes en el grupo de ruxolitinib en comparación con el 23,5% en el grupo de MTD; el aumento correspondiente en casos de grado 3 y 4 fue similar (10,6% en comparación con 6,2%). Durante el seguimiento extendido en pacientes que recibieron ruxolitinib, los valores de lipasa aumentada se produjeron en el 35,9% de los pacientes; la enfermedad de grado 3 y 4 se produjo en el 9,5% y el 0,4% de los pacientes, respectivamente.
Aumento de la presión arterial sistólica
En los estudios de fase 3 en pacientes con MF, el aumento de la presión arterial sistólica desde el nivel basal en 20 mmHg o más en al menos una de las visitas se documentó en el 31,5% de los pacientes en comparación con el 19,5% de los pacientes que recibieron tratamiento de control. En el estudio COMFORT I (pacientes con MF), el aumento medio de la presión arterial sistólica desde el nivel basal fue de 0-2 mmHg con ruxolitinib en comparación con una disminución de 2-5 mmHg en el grupo de placebo. Los resultados del estudio COMFORT II mostraron una diferencia mínima en los valores medios entre pacientes con MF que recibieron ruxolitinib y aquellos que recibieron tratamiento de control.
En el período aleatorizado del estudio en pacientes con PV, el aumento medio de la presión arterial sistólica aumentó en 0,65 mmHg en el grupo de ruxolitinib en comparación con una disminución de 2 mmHg en el grupo de tratamiento óptimo disponible.
Niños y adolescentes
Un total de 20 pacientes con edades comprendidas entre 12 y <18 años con TTP se analizaron para la seguridad: 9 pacientes (5 en el grupo de ruxolitinib y 4 en el grupo de MTD) en el estudio REACH2 y 11 pacientes (4 en el grupo de ruxolitinib y 7 en el grupo de MTD) en el estudio REACH3. Considerando la exposición similar observada en niños y adultos, las reacciones adversas a ruxolitinib con la dosis recomendada de 10 mg dos veces al día son similares en frecuencia y gravedad.
Pacientes de edad avanzada
Un total de 29 pacientes en el estudio REACH2 y 25 pacientes en el estudio REACH3 con edades > 65 años que recibieron ruxolitinib se analizaron para la seguridad. No se encontraron nuevos problemas de seguridad, y el perfil de seguridad en pacientes > 65 años es generalmente consistente con el perfil de seguridad observado en pacientes de 18 a 65 años.
Notificación de sospechas de reacciones adversas
La notificación de reacciones adversas después de la autorización del medicamento es importante. Esto permite realizar un seguimiento de la relación beneficio/riesgo del medicamento. Los profesionales sanitarios y los pacientes o sus representantes legales deben notificar todas las sospechas de reacciones adversas y falta de eficacia del medicamento a través del Sistema de Farmacovigilancia en https://aisf.dec.gov.ua
Fecha de caducidad
3 años.
Condiciones de conservación
Conservar a una temperatura no superior a 25 °C. Conservar en un lugar inaccesible para los niños.
Envase
14 tabletas en blister, 4 blisters en caja de cartón para envase.
Categoría de prescripción
Con receta.
Fabricante
Novartis Pharma Stein AG.
Dirección del fabricante
Schaffhauserstrasse, 4332 Stein, Suiza.
- País de registro
- Requiere recetaNo
- Fabricante
- Esta información ha sido traducida con IA y es solo orientativa. No constituye asesoramiento médico. Consulta siempre con un médico antes de tomar cualquier medicamento.
- Alternativas a RENNI Z APEL'SINOVIM SMAKOMForma farmacéutica: suspensión, 170 ml en una botellaPrincipio activo: ordinary salt combinationsFabricante: Балканфарма-Троян АТNo requiere recetaForma farmacéutica: suspensión, 170 ml en una botella; 10 ml en un sobrePrincipio activo: combinationsFabricante: Балканфарма-Троян АТNo requiere recetaForma farmacéutica: tabletas, 6 tabletas en una ampollaPrincipio activo: ordinary salt combinationsFabricante: ПЛІВА Хрватска д.о.о.No requiere receta
Alternativas a RENNI Z APEL'SINOVIM SMAKOM en otros países
Las mejores alternativas con el mismo principio activo y efecto terapéutico.
Alternativa a RENNI Z APEL'SINOVIM SMAKOM en Polonia
Médicos online para RENNI Z APEL'SINOVIM SMAKOM
Consulta sobre dosis, efectos secundarios, interacciones, contraindicaciones y renovación de la receta de RENNI Z APEL'SINOVIM SMAKOM – sujeta a valoración médica y normativa local.









Mantente informado y ahorra en salud
Recibe consejos de salud, novedades de la plataforma y promociones exclusivas para suscriptores.

