

OBIZUR 500 U POLVO Y DISOLVENTE PARA SOLUCION INYECTABLE




Cómo usar OBIZUR 500 U POLVO Y DISOLVENTE PARA SOLUCION INYECTABLE
Traducción generada por IA
Este contenido ha sido traducido automáticamente y se ofrece solo con fines informativos. No sustituye la consulta con un profesional sanitario.
Ver originalContenido del prospecto
Introducción
Prospecto: información para el usuario
OBIZUR500U polvo y disolvente para solución inyectable
Susoctocog alfa
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
- Qué es OBIZUR y para qué se utiliza
- Qué necesita saber antes de empezar a usar OBIZUR
- Cómo usar OBIZUR
- Posibles efectos adversos
- Conservación de OBIZUR
- Contenido del envase e información adicional
1. Qué es OBIZUR y para qué se utiliza
OBIZUR contiene el principio activo susoctocog alfa, factor VIII antihemofílico, secuencia porcina. El factor VIII es necesario para que la sangre forme coágulos y para detener los sangrados.
En los pacientes con hemofilia adquirida, el FVIII no funciona correctamente porque el paciente ha producido anticuerpos contra su propio factor VIII que neutralizan este factor de la coagulación sanguínea.
OBIZUR se utiliza para el tratamiento de los episodios de sangrado en los adultos con hemofilia adquirida (un trastorno hemorrágico causado por la falta de actividad de factor VIII debido a la producción de anticuerpos). El efecto neutralizador de estos anticuerpos frente a OBIZUR es menor que frente al factor VIII humano.
OBIZUR restablece la actividad de factor VIII ausente y ayuda a que la sangre forme coágulos en el lugar del sangrado.

2. Qué necesita saber antes de empezar a usar OBIZUR
El producto solo se puede administrar a los pacientes hospitalizados, ya que es necesario supervisar clínicamente el estado hemorrágico del paciente.
No use OBIZUR:
- si es alérgico al susoctocog alfa o a alguno de los demás componentes de este medicamento (incluidos en la sección 6)
- si es alérgico a las proteínas de hámster (OBIZUR puede contener cantidades mínimas derivadas del proceso de fabricación)
En caso de duda, consulte a su médico antes de empezar a usar este medicamento.
Advertencias y precauciones
Consulte a su médico antes de empezar a usar OBIZUR.
Existe una posibilidad muy pequeña de que sufra una reacción alérgica a OBIZUR. Debe estar al tanto de los signos iniciales de las reacciones alérgicas (ver sección 4 para consultar los signos y síntomas). Si aparece alguno de estos síntomas, se debe detener la inyección. Los síntomas graves, como la dificultad para respirar y el (pre)desmayo, necesitan tratamiento urgente.
Pacientes que producen anticuerpos inhibidores contra OBIZUR
Su médico puede comprobar su tiene anticuerpos inhibidores contra el factor VIII porcino.
Su médico comprobará el factor VIII en la sangre para confirmar que se le está administrando suficiente factor VIII. Su médico también comprobará si se ha detenido satisfactoriamente el sangrado.
Informe a su médico si ha tenido una enfermedad cardiovascular en el pasado o si tiene riesgo conocido de trombosis (enfermedades producidas por coágulos de sangre en los vasos sanguíneos normales), ya que no se puede descartar la posibilidad de sufrir enfermedades tromboembólicas con la administración de concentraciones altas y prolongadas de factor VIII.
Nombre y número de lote
Recomendamos encarecidamente que el profesional médico registre el nombre y el número de lote del medicamento cada vez que se use OBIZUR, con el fin de mantener un vínculo entre su tratamiento y el lote del medicamento.
Niños y adolescentes
OBIZUR no está autorizado actualmente para el tratamiento de pacientes menores de 18 años de edad, en los que la hemofilia adquirida es rara.
Uso de OBIZUR con otros medicamentos
Informe a su médico si está utilizando, ha utilizado recientemente o pudiera tener que utilizar cualquier otro medicamento. No se conocen interacciones entre OBIZUR y otros medicamentos.
Embarazo y lactancia
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico antes de utilizar este medicamento.
Conducción y uso de máquinas
OBIZUR no influye en la capacidad para conducir y utilizar máquinas.
OBIZUR contiene sodio
Este medicamento contiene 4,4 mg de sodio por mililitro
una vez preparado.
Consulte a su médico si sigue una dieta pobre en sodio.
3. Cómo usar OBIZUR
El tratamiento con OBIZUR lo llevará a cabo un médico con experiencia en la atención de pacientes con hemofilia (trastornos hemorrágicos).
Su médico calculará la dosis de OBIZUR (en unidades o U) dependiendo de su estado y peso corporal. La frecuencia y la duración de la administración dependerán del grado de eficacia que tenga OBIZUR en su caso. Normalmente, el tratamiento sustitutivo con OBIZUR es temporal hasta que desaparezca el sangrado o se erradiquen los anticuerpos contra su propio factor VIII.
La dosis de inicio recomendada es de 200 U por kilogramo de peso corporal administradas mediante inyección intravenosa.
Su médico analizará su actividad de factor VIII cada cierto tiempo para decidir la siguiente dosis y frecuencia de OBIZUR.
Lo habitual es que el sangrado remita en las primeras 24 horas; su médico ajustará la dosis y la duración de OBIZUR hasta que deje de sangrar.
El volumen total de OBIZUR reconstituido se debe administrar a una velocidad de entre 1 y 2 ml por minuto.
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico.
Si usa más OBIZUR del que debe
Siga exactamente las instrucciones de administración de OBIZUR indicadas por su médico. Si usa más OBIZUR del recomendado, informe a su médico lo antes posible.
Si olvidó usar OBIZUR
No use una dosis doble para compensar las dosis olvidadas. Consulte a su médico si ha olvidado una dosis y no sabe cómo compensarlo.
Si interrumpe el tratamiento con OBIZUR
No interrumpa el tratamiento con OBIZUR sin consultar a su médico.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico.

4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Si se producen reacciones alérgicas graves y repentinas, se debe suspender la inyección inmediatamente. Contacte con su médico inmediatamente si tiene alguno de los siguientes síntomas iniciales:
- Hinchazón de labios y lengua
- Escozor y punzada en el lugar de inyección
- Escalofrío, enrojecimiento
- Habón urticarial, picor generalizado
- Dolor de cabeza, presión arterial baja
- Adormecimiento (letargo), sensación de enfermedad, inquietud
- Latido rápido del corazón, opresión en el pecho
- Cosquilleo, vómitos
- Sonido silbante que se produce al respirar (sibilancia)
Efectos adversos frecuentes (pueden afectar hasta1persona de cada10)
- Producción de anticuerpos contra el medicamento
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de OBIZUR
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en la caja, el vial y la jeringa precargada después de CAD. La fecha de caducidad es el último día del mes que se indica.
Conservar en nevera (entre 2ºC y 8ºC).
No congelar.
Utilice la solución reconstituida inmediatamente y nunca más de 3 horas después de que el polvo se haya disuelto por completo.
Tras la reconstitución, la solución debe ser transparente e incolora.
No la administre si detecta partículas o cambio de color.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Titular de la autorización de comercialización y responsable de la fabricación
Composición de OBIZUR
- El principio activo es susoctocog alfa (factor VIII antihemofílico, secuencia porcina, producido mediante tecnología de DNA recombinante). Cada vial de polvo contiene 500 de U susoctocog alfa.
- Los demás componentes del polvo son polisorbato 80, cloruro de sodio (ver también la sección 2), cloruro de calcio dihidratado, sacarosa, Tris base, Tris HCl, citrato trisódico dihidratado.
- El disolvente es 1 ml de agua esterilizada para preparaciones inyectables.
Aspecto del producto y contenido del envase
Un envase contiene 1, 5 o 10 unidades de lo siguiente:
- vial de vidrio con 500 U de OBIZUR en forma de polvo friable blanco provisto de tapón de caucho y cápsula de cierre de tipo «flip‑off»
- jeringa de vidrio precargada con 1 ml de agua esterilizada para preparaciones inyectables provista de protector de caucho butílico y adaptador para conexión Luer
- dispositivo de transferencia de líquido con punzón de plástico integrado
Titular de la autorización de comercialización y responsable de la fabricación
Titular de la autorización de comercialización
Baxalta Innovations GmbH
Industriestrasse 67
A‑1221 Viena
Austria
Responsable de la fabricación
Baxter AG
Industriestrasse 67
A‑1221 Viena
Austria
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
België/Belgique/Belgien Baxalta Belgium SPRL Tél./Tel.: +32 2 892 62 00 | Lietuva UAB Baxter Lithuania Tel: +370 5 269 16 90 / +370 5 252 71 00 |
???????? ??????? ???????? ???? ???.: + 359 2 9808482 | Luxembourg/Luxemburg Baxalta Belgium SPRL Tél/Tel: +32 2 892 62 00 |
Ceská republika Baxter Czech spol.s r.o. Tel.: +420 225774111 | Magyarország Baxter Hungary Kft Tel.: +36 1 202 1980 |
Danmark Baxalta Denmark A/S Tlf.: +45 32 70 12 00 | Malta Baxalta UK Limited Tel.: +44 1 635 798 777 |
Deutschland Baxalta Deutschland GmbH Tel.: +49 89 262077‑011 | Nederland Baxalta Netherlands B.V. Tel.: +31 30 799 27 77 |
Eesti OÜ Baxter Estonia Tel.: +372 6 515 120 | Norge Baxalta Norway AS Tlf.: +47 22 585 000 |
Ελλ?δα Baxter Hellas ΕΠΕ Τηλ.: +30 210 28 80 000 | Österreich Baxalta Österreich GmbH Tel.: +43 1 20100‑0 |
España Baxalta Spain S.L. Tel.: +34 91 790 42 22 | Polska Baxter Polska Sp. z.o.o. Tel.: +48 22 4883 777 |
France Baxalta France SAS Tél.: +33 1 70 96 06 00 | Portugal Baxalta Portugal, Unipessoal, Lda. Tel.: +351 21 122 03 00 |
Hrvatska Baxter d.o.o. Tel.: +386 1 420 16 80 | România FARMACEUTICA REMEDIA SA Tel.: + 40 21 321 16 40 |
Ireland Baxalta UK Limited Tel.: +44 1 635 798 777 | Slovenija Baxter d.o.o. Tel.: +386 1 420 16 80 |
Ísland Lyfjaver ehf. Sími: +354 533 6100 | Slovenská republika Baxter Slovakia, s.r.o. Tel: +421 2 3210 1150 |
Italia Baxalta Italy S.r.l. Tel.: +39 06 45224 600 | Suomi/Finland Baxalta Finland Oy Puh/Tel.: +358 201 478 200 |
Κ?προς Baxter Hellas ΕΠΕ Τηλ.: +30 210 28 80 000 | Sverige Baxalta Sweden AB Tel.: +46 8 50 53 26 00 |
Latvija SIA Baxter Latvia Tel.: +371 67 784 784 | United Kingdom Baxalta UK Limited Tel.: +44 1 635 798 777 |
Fecha de la última revisión de este prospecto:
Este medicamento se ha autorizado en «circunstancias excepcionales». Esta modalidad de aprobación significa que debido a la rareza de su enfermedad no ha sido posible obtener información completa de este medicamento.
La Agencia Europea de Medicamentos revisará anualmente la información nueva de este medicamento que pueda estar disponible y este prospecto se actualizará cuando sea necesario.
Otras fuentes de información
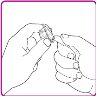
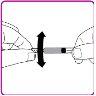
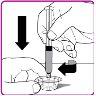
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu <, y en la página web de la {Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) (http://www.aemps.gob.es/)}>. ‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑ Esta información está destinada únicamente a profesionales del sector sanitario: INSTRUCCIONES PARA LA PREPARACIÓN Y ADMINISTRACIÓN Preparación Antes de comenzar la reconstitución, necesitará lo siguiente: Los siguientes procedimientos son directrices generales para la preparación y reconstitución de OBIZUR. Repita las siguientes instrucciones de reconstitución con cada vial de polvo que vaya a reconstituir. Reconstitución Utilice una técnica aséptica durante el procedimiento de reconstitución. Figura A Figura B Figura C Figura D Figura E Figura F Figura G Figura H Administración ¡Solo para inyección intravenosa! Utilizando una técnica aséptica, administre la solución siguiendo el siguiente procedimiento: Figura I La dosis inicial de OBIZUR necesaria para un paciente se calcula mediante la siguiente fórmula: Dosis inicial (U/kg) ? concentración del producto (U/vial) × peso corporal (kg) = número de viales por ejemplo, el número de viales para la dosis inicial en un sujeto de 70 kg se calculará de la forma siguiente: 200 U/kg ? 500 U/vial × 70 kg = 28 viales Dosificación La dosis inicial recomendada es de 200 U por kilogramo de peso corporal, administradas mediante inyección intravenosa. Tipo de sangrado Actividad mínima de factor VIII deseada (Unidades por dl o % de lo normal) Dosis inicial (Unidades por kg) Dosis siguiente Frecuencia y duración de las dosis siguientes Sangrado leve a moderado del músculo superficial/sin afectación neurovascular y sangrado articular > 50 % 200 Ajuste las dosis siguientes en función de la respuesta clínica y para mantener la actividad mínima deseada de factor VIII Administre las dosis a intervalos de entre 4 y 12 horas; la frecuencia puede ajustarse en función de la respuesta clínica y la actividad de factor VIII cuantificada Sangrado intramuscular, retroperitoneal, gastrointestinal, intracraneal importante de moderado a grave > 80 %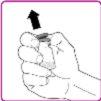
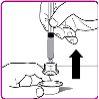
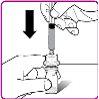
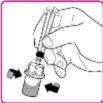
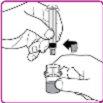
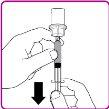
- País de registro
- Principio activo
- Requiere recetaSí
- Fabricante
- Esta información es de carácter general y no sustituye la consulta con un profesional sanitario.
- Alternativas a OBIZUR 500 U POLVO Y DISOLVENTE PARA SOLUCION INYECTABLEForma farmacéutica: INYECTABLE, 1.000 UIPrincipio activo: factor de coagulación VIIIFabricante: Takeda Manufacturing Austria AgRequiere recetaForma farmacéutica: INYECTABLE, 1.500 UIPrincipio activo: factor de coagulación VIIIFabricante: Takeda Manufacturing Austria AgRequiere recetaForma farmacéutica: INYECTABLE, 1000 UI - tras reconstitución en 2 ml de agua para inyectables la dosis es de 500 UI/mlPrincipio activo: factor de coagulación VIIIFabricante: Takeda Manufacturing Austria AgRequiere receta
Médicos online para OBIZUR 500 U POLVO Y DISOLVENTE PARA SOLUCION INYECTABLE
Comenta la dosis, los posibles efectos secundarios, interacciones, contraindicaciones o la revisión de receta de OBIZUR 500 U POLVO Y DISOLVENTE PARA SOLUCION INYECTABLE, sujeto a valoración médica y a la normativa local.









Preguntas frecuentes
